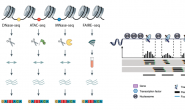
بسم الله الرحمن الرحيم
في المقال السابق شرحنا المبدأ الذي تقوم عليه تقنية الـ Microarray وأنها تقوم بتلوين الـ mRNA أو الـ cDNA لكل نوع من الخلايا باللون الأحمر أو الأخضر وبعدها تُقارَن نسبة الإختلاف بين نسبة إشعاع الجين في الحالتين ونقوم بإختيار الجينات التي أظهرت إختلافا ملحوظا بين العينات. ورأينا أن هناك نوعين من هذه الرقائق, رقائق تصنع باستخدام الروبوت (Spotted Array) ورقائق تصنع باستعمال أقنعة وأشعة ضوئية باستعمال طريقة In-sito spotting , والمتمثلة العادة في رقائق Affymetrix , يسمى هذا النوع من الرقائق بالـ oligonucleotides ships.
مالم نقله في المقال السابق هو أنه خلافا للرقائق المصنعة بإستعمال الروبوت, يتم إستعمال نوع واحد من الخلايا في كل تجربة في شرائح Affymetrix, أي يعني أنه يجب القيام بتجربتين (واحدة باستعمال الخلايا العادية وأخرى باستعمال الخلايا المريضة). لهذا تطلق تسمية الرقائق ثنائية القناة (Dual channel array) على الرقائق التي تسمح باستعمال لونين في نفس الوقت و تسمية رقائق أحادية القناة (Single channel array) على الرقائق التي تسمح باستعمال لون واحد.
في هذا المقال سوف نركز على طرق تحليل هذه البيانات وما هي الإحتياطات التي يجب أخذها بعين الإعتبار في كل مرحلة. في العادة يتم تقسيم مراحل القيام بتجارب الـ Microarray إلى سبعة مراحل أساسية كما هو موضح في الشكل.

مخطط بياني يوضح أهم مراحل تجارب الـ Microarray
1. السؤال البيولوجي:
حتى وإن كانت هذه المرحلة بديهية لكنها في رأيي هي من أهم المراحل إذا يجب على الباحث قبل الشروع في التجربة, معرفة نوع المشكلة التي يريد الإجابة عنها, مثلا: ماهو نوع المرض ؟ ماهي خصائص العينات التي يجب أخذها؟ ما هي الحالة التي يجب أن تكون العينات فيها؟ .... إلخ. تحديد الهدف في هذه المرحلة يساهم في زيادة إحتمال الحصول على النتائج المرجوة ومن ثم فهم نتائج التجربة وتصحيح الأخطاء إن وجدت.
2.تصميم التجربة:
يتم خلال هذه المرحلة التفكير في تفاصيل التجربة و تحضير العينات. مع إنخفاض سعر الرقائق يستحسن إستعمال عدة عينات من النوعين (مثلا عشرة عينات لخلايا سليمة و عشرة عينات مريضة) من أجل زيادة الدقة الإحصائية و إلغاء الأخطاء الموجودة في كل عينة, حيث من المعلوم أنه لو أعدنا التجربة بإستعمال نفس العينة ونفس الكمية ونستعمل نفس الجهاز ويقوم بالتجربة نفس الشخص لن تكون النتائج مطابقة 100% .
كما يجب التنويه أيضا أن هناك نوعين من العينات المكررة (Replicates):
- المتكررات التقنية (Technical Replicate) : وهي عبارة عن نسخ تُأخذُ من نفس العينة البيولوجية. مثلا نأخذ عينة من مريض واحد بكمية كبيرة نوعا ما ونقوم بقسيمها إلى كميات متساوية ونقوم بالتجربة في كل مرة بإستخدام واحدة من النسخ. كما يمكن أيضا وضع نُسَخ عدة من cDNA الجين في نفس الشريحة. تسمح لنا المتكررات التقنية بمعرفة نسبة الخطأ على مستوى نوع واحد من العينتين.
- المتكررات البيولوجية (Biological Replicates): وهي نسخ مأخوذة من مصادر مختلفة لنفس الحالة. مثلا يتم أخذ ثلاث عينات لخلايا عادية من ثلاثة متطوعين أصحاء. من الجانب الإحصائي تسمح لنا المتكرارت البيولوجية من معرفة نسبة الخطأ على مستوى مجتمع العينات.
بالإضافة إلى هذا يجب أيضا تحديد نوع الرقائق التي سوف تُستعمل, هل يجب إستعمال رقائق ثنائية القناة أم رقائق أحادية القناة. في حالة إستعمال رقيقة ثنائية القنات يجب فقط الأخذ بعين الإعتبار أن نسبة التهجين و نسبة الـ mRNA ستكون مختلفة بين العينتين وحتى إن كانت النسبة متساوية فإن نسبة إشعاع اللون الأحمر (Cy3) و اللون الأخضر (Cy5) ليست متساوية. لتصحيح هذه المشكلة نقوم بعملية قلب الألوان, حيث نقوم بالتجربة مرتين وفي كل تجربة نقلب الألوان بين العينتن, فالعينة التي كانت ملونة بالأحمر في التجربة الألى تلون بالأخضر والعينة التي لُوِنت بالإخضر في التجربة الأولى تلون بالأحمر وتعاد التجربة.

رسم تخطيطي يوضح مبدأ قلب الألوان
3. القيام بتجربة الـ Microarray:
بعد تجهيز كل العينات وإختيار نوع الرقيقة نقوم بالتجربة.
4. معالجة الصور وحساب نسبة النشاط:
في هذه المرحلة تستعمل برامج متخصصة من أجل تحديد نسبة الإشعاع في كل مسبار وتحديد نسبة التشويش الناجمة عن الإشعاعات الناجمة عن مصادر أخرى غير عملية التهجين. إذ أن هناك عدة عوامل يمكن أن تأثر على نسبة الإشعاع كوضعية الرقيقة, ضغط الغرفة,... إلخ. لهذا يجب أولا حذف هذه التشويشات الخلفية (Background Signal) والإحتفاظ بالإشارة الحقيقية قدر الإمكان. تختلف البرامج المستعملة في تحليل الصور لكنها كلها تعمل على حسب مبدأ واحد هو حساب نسبة الإشعاع في النقطة الأكثر كثافة بالنسبة لكل مسبار . بعد ذالك يتم حذف نسبة التشويش. ليكن 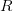 نسبة الإشعاع الأحمر الأولية وليكن
نسبة الإشعاع الأحمر الأولية وليكن  نسبة الإشعاع الأخضر الأولية. بعد حساب نسبة التشويشات الخلفية
نسبة الإشعاع الأخضر الأولية. بعد حساب نسبة التشويشات الخلفية 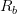 بالنسبة للأحمر و
بالنسبة للأحمر و  بالنسبة للأخضر يتم حساب نسبة نشاط (تعبير) الجين بحذف نسبة التشويش فتصبح نسبة الإشعاع تساوي
بالنسبة للأخضر يتم حساب نسبة نشاط (تعبير) الجين بحذف نسبة التشويش فتصبح نسبة الإشعاع تساوي 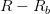 بالنسبة للأحمر و
بالنسبة للأحمر و 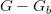 بالنسبة للأخضر ويمكننا حساب نسية التغيير
بالنسبة للأخضر ويمكننا حساب نسية التغيير 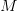 كالتالي:
كالتالي:
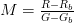
هذه الطريقة تعتبر بسيطة لكن من بعض مشاكلها أنها تؤدي إلى قيم سالبة. طورت طرق أخرى للتخلص من نسبة التشويش أكثر فعالية.

صورة توضح مبدا عمل بعض برامج معالجة الصور (المصدر)
بالنسبة لرقائق أحادية القناة يختلف تصميم الرقيقة حيث يتم تصميم مسبارين لكل جين. المسبار الأول يحتوي على سلسلة مطابقة لسلسلة الجين ( Perfect Match) ونرمز لها بـ 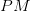 وسلسلة مغايرة (Missmatch) ونرمز لها بـ
وسلسلة مغايرة (Missmatch) ونرمز لها بـ 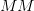 , المبدأ هنا هو أن الإشعاع الناتج عن عملية التهجين في المسابير ذات السلسلة المطابقة تمثل نسبة النشاط الحقيقي والإشعاع الناتج عن السلسلة المغايرة يمثل التشويش والهدف أن يكون الفرق بين هذين الإشعاعين كبير, فإذا كان متساوي فهذا يعني أن القياس غير صحيح. في العادة تقاس نسبة الفرق بين الإشعاعين كمتوسط التغير بين كل ثنائي مسابير بستعمال العبارة التالية
, المبدأ هنا هو أن الإشعاع الناتج عن عملية التهجين في المسابير ذات السلسلة المطابقة تمثل نسبة النشاط الحقيقي والإشعاع الناتج عن السلسلة المغايرة يمثل التشويش والهدف أن يكون الفرق بين هذين الإشعاعين كبير, فإذا كان متساوي فهذا يعني أن القياس غير صحيح. في العادة تقاس نسبة الفرق بين الإشعاعين كمتوسط التغير بين كل ثنائي مسابير بستعمال العبارة التالية
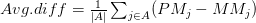
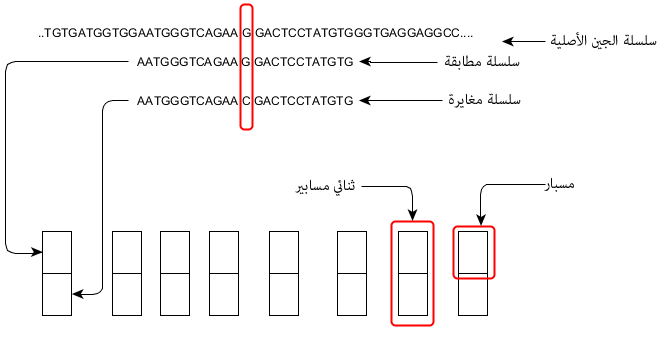
رسم توضيحي يبين مبدأ الرقائق أحادية القناة
5. مقايس الجودة:
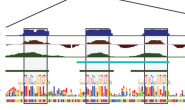
بعد حساب نسبة إشعاع كل جين وحساب قيمة التشويش الخلفي يستحسن القيام بقياس نسبة جودة البيانات لمعرفة هل يمكن مواصلة إستعمالها ومعرفة أيضا مدى نسبة نجاح التجربة. لاتوجد بروتوكولات محددة من أجل تحديد نسبة الجودة, لكن من القياسات المستعملة بكثرة هي قياس نسبة الإشارة إلى الضجيج (Signal to Noise Ratio أو SNR) والتي كلما كانت كبيرة كانت الجودة جيدة. هناك عدة طرق لحساب هذه القيمة ويمكن التطلع عليها في مصادر أخرى. مثلا في البيان الموضع في الصورة في الأسفل يوضح رسم بياني لقيمة SNR للإشارتين الحمراء والخضراء لتجربتين. نلاحظ أن نسبة الـ SNR في التجربة الممثلة في الصورة العلوية أحسن من الصورة العلوية لأن نسبة الإشارة الحقيقية أكبر من نسبة الضجيج إما في الثانية نلاحظ أنها ضعيفة.
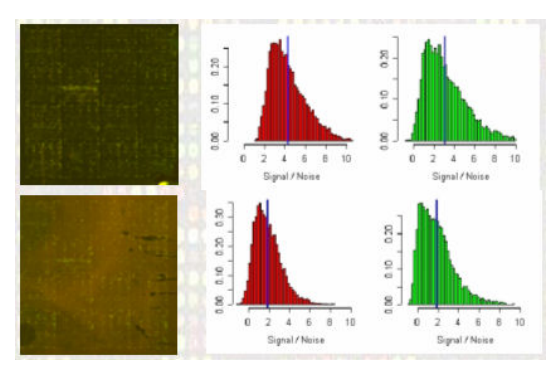
مقارنة نسبة الإشارة إلى الضجيج لعينتين (المصدر)
6. Normalization:
تعتبر هذه المرحلة مرحلة حساسة في معالجة بيانات الـ Microarray, حيث أن تنائجها هي التي تحدد دقة النتائج النهائية. كما قلنا سابقا أن هناك دائما إختلاف بين القياسات لتجربتين مختلفتين وذالك ناتج عن أسباب تقنية. فعند المقارنة بين عينتين يجب أن تكون القيم قابلة للمقارنة, فمثلا في التجربة الأول كانت الكمية المستعملة كبيرة وفي الثانية كانت أقل بالطبع سوف تؤدي إلى قيم مختلفة في الإشعاع لكن هذا لا يعني أن جينات التجربة الأولى أكثر نشاطا من التجربة الثانية لهذا وجبت الـ Normalization (تنسيب القيم إلى الواحد).
تعتمد الطرق المستعملة في الـ Normalization على أن فقط نشاط بعض الجينات يتغير بين عينتين وهي الجينات المسؤولة عن الإختلاف أما أغلبية الجينات فلا يكون هناك تغير ملحوض في نسبة نشاطها. لهذا تم تصميم بعض الرقائق لتحتوي على مجموعة من الجينات تسمى الجينات المحافظة (Housekeeping genes) وهي جينات يعتقد أن نسبة نشاطها ثابتة بين كل أنواع الخلايا وبالتالي يمكننا إستعمالها لتوحيد قيمة النشاط بين عينتين. لكن هذه الطريقة لها بعض السلبيات.
من بين الطرق الأكثر إستعمالا أيضا هي طريقة الـ Normalization بإستعمال طريقة الإنحدار (Regression) الموضعي (لست متأكد من الترجمة) LOWESS . تقوم هذه الطريقة على الملاحظة القائمة أن نسبة الإشعاع بين الملون الكيميائي الأحمر والأخضر تختلف على حسب نسبة تركيز المحلول وأن المكونات الكيميائة تمتص إشعاع المكونات المجاورة مما يؤدي إلى نوع من الإنحياز في نوع اللون النشط (Dye Bias). فالأصل أننا إذا قمنا بمقارنة نسبة إشعاع اللون الأحمر واللون الأخضر فإن النسبة بينهما  ستكون مساوية للواحد بالنسبة لأغلب الجينات, يعني لورسمناها في بيان الإنتشار (Scatter plot) ستكون أغلبها على خط المحور 45 درجة.
ستكون مساوية للواحد بالنسبة لأغلب الجينات, يعني لورسمناها في بيان الإنتشار (Scatter plot) ستكون أغلبها على خط المحور 45 درجة.
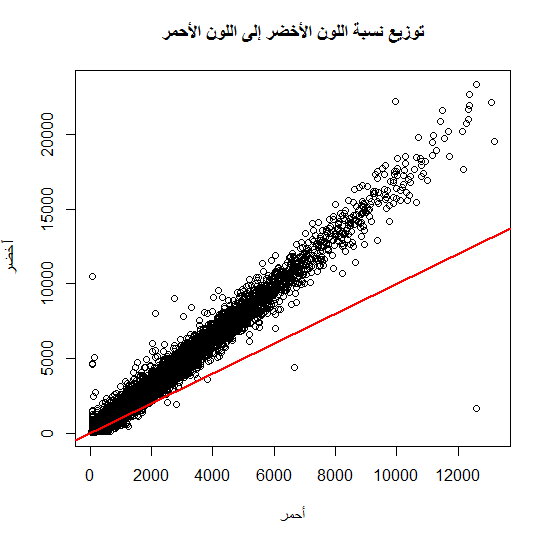
بيان إنتشار توزيع إشعاع اللون الأحمر بالنسبة للون الأخضر, الخط الأحمر يمثل خط 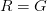
لكن في أغلب الأحيان لا نلاحظ هذا التوزيع حيث تكون النقاط على شكل فاكهة الموز (Banana Shape) وهذا يعني أنه يجب علينا تصحيحها. من الطرق المستعملة جدا هي رؤية البيانات بإستعمال طريقة MA plot, حيث نسبة الإشعاع الاحمر والأخضر $M$ لكل جين مع متوسط التغير بين الإشعاعين $A$. في الحقيقة لا يمثل هذا البيان إلا إدارة بنسبة 45 درجة للبيان الذي يقارن اللونين الأحمر والأخضر. يتم حساب قيمة و 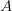 كالتالي:
كالتالي:


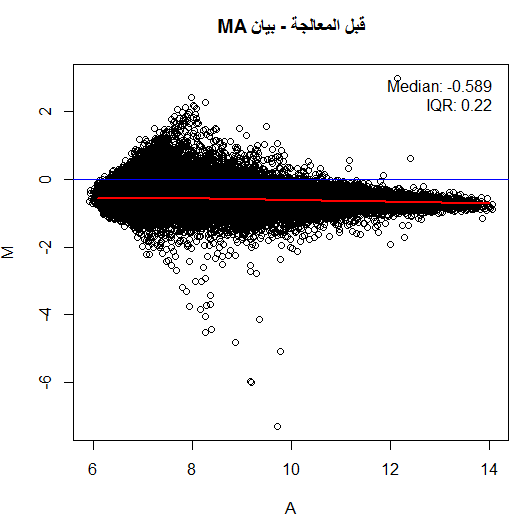
توضيح مبدأ بيان MA, اللون الأحمر يمثل منحنى التوفيق والأزرق المبدأ
لتصحيح قيمة البيانات هنا يجب علينا أن نرجع أغلبية النقاط إلى الخط 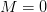 بحيث تكون أغلبية الجينات لها نفس النشاط تقريبا بين العينتين. للقيام بذالك ماعلينا إلا أن نقوم إيجاد منحني يمر عبر أغلبية النقط (عملية توفيق المنحني, Curve fitting) ثم نقوم بحذف هذه القيمة من القيمة الحقيقية وبالتالي نقوم بإصعاد (أو إنزال) النقاط إلى خط . تتم عملية توفيق المنحنى باستعمال طريقة تسمى LOWESS وهي طريقة إنحدار تقوم بتقسيم البيان إلى مجالات صغيرة و توفيق منحنى على كل جزء. يمكن كتابة العملية بالمعادلة التالية:
بحيث تكون أغلبية الجينات لها نفس النشاط تقريبا بين العينتين. للقيام بذالك ماعلينا إلا أن نقوم إيجاد منحني يمر عبر أغلبية النقط (عملية توفيق المنحني, Curve fitting) ثم نقوم بحذف هذه القيمة من القيمة الحقيقية وبالتالي نقوم بإصعاد (أو إنزال) النقاط إلى خط . تتم عملية توفيق المنحنى باستعمال طريقة تسمى LOWESS وهي طريقة إنحدار تقوم بتقسيم البيان إلى مجالات صغيرة و توفيق منحنى على كل جزء. يمكن كتابة العملية بالمعادلة التالية:
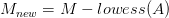
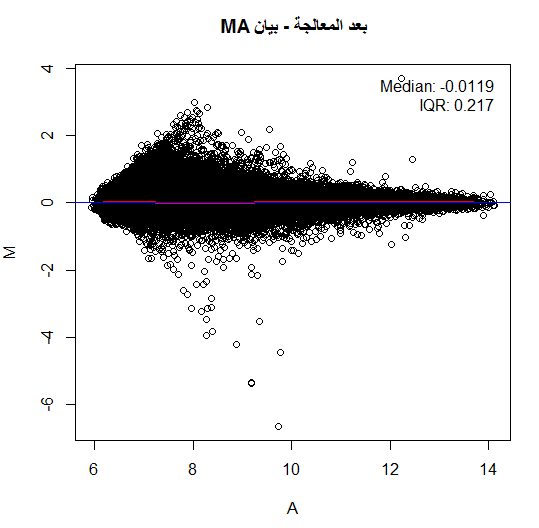
تصيح البيانات في بيان MA
7. تحليل نشاط الجينات:
في المراحل السابقة قمنا بمحاولة تصحيح القيم المتحصل عليها والتخلص من أية تشويشات تقنية. في هذه المرحلة نستعمل هذه البيانات التي نظن أنها جيدة في محاولة إيجاد مجموعة الجينات التي طرء عليها تغير ملموس في قيمة نشاطها (Differently expressed Genes).
لنفرض أنى لدينا 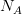 عينة جيدة و
عينة جيدة و 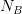 عينة مريضة وليتكن نسبة نشاط (تعبير) كل جين هي
عينة مريضة وليتكن نسبة نشاط (تعبير) كل جين هي 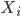 بالنسبة للجين
بالنسبة للجين 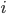 في المجموعة الأولى و
في المجموعة الأولى و 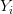 بالنسبة للجين في المجموعة الثانية. الطريقة السهلة هي حساب القيمة المطلقة للوغرتم نسبة التغير بين متوسط نشاط الجين $i$ في العينات الغير مريضة و متوسط نشاط الجين في العينات المريضة و نرتب الجينات من الجينات ذات أكبر تغير إلى الأصغر وتسمى هذه الطريقة, طريقة (الـ Fold-Change). نلاحظ أن القيمة المطلقة تسمح لنا بالأخذ بعين الإعتبار الجينات التي زادت في نسبة التعبير (Up-regulateg Genes) و الجينات التي نقصت (Down-regulated Genes). ويمكننا كتابتها بالشكل التالي:
بالنسبة للجين في المجموعة الثانية. الطريقة السهلة هي حساب القيمة المطلقة للوغرتم نسبة التغير بين متوسط نشاط الجين $i$ في العينات الغير مريضة و متوسط نشاط الجين في العينات المريضة و نرتب الجينات من الجينات ذات أكبر تغير إلى الأصغر وتسمى هذه الطريقة, طريقة (الـ Fold-Change). نلاحظ أن القيمة المطلقة تسمح لنا بالأخذ بعين الإعتبار الجينات التي زادت في نسبة التعبير (Up-regulateg Genes) و الجينات التي نقصت (Down-regulated Genes). ويمكننا كتابتها بالشكل التالي:
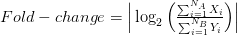
من سلبيات هذه الطريقة أنها لا تأخذ بعين الإعتبار التغير بين العينات لهذا في العادة نقوم بالإختبار الإحصائي "تي" (T-test) والذي يهدف إلى معرفة هل نسبة التغير بين متوسط نشاط الجين بين النوعين ملموس أم لا, وفي العاد تحسب كالآتي:
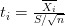
بحيث تمثل 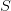 الإنحراف المعياري و
الإنحراف المعياري و 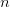 عدد العينات. هذه الطريقة تكون في العادة لكن بما أنه في أغلب الحالات يكون عدد العينات قليلا يتم تصحيح الإنحراف المعياري بإستعمال طريقة الـ variance Shrinkage لزيادة دقة الإنحراف المعياري بإستخدام المعلومات عن الجينات الأخرى في مختلف العينات. من الطرق المستعملة بكثرة هي الطريقة المستعملة في حزمة limma بلغة الـ R والتي تحسب كالتالي:
عدد العينات. هذه الطريقة تكون في العادة لكن بما أنه في أغلب الحالات يكون عدد العينات قليلا يتم تصحيح الإنحراف المعياري بإستعمال طريقة الـ variance Shrinkage لزيادة دقة الإنحراف المعياري بإستخدام المعلومات عن الجينات الأخرى في مختلف العينات. من الطرق المستعملة بكثرة هي الطريقة المستعملة في حزمة limma بلغة الـ R والتي تحسب كالتالي:
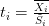
بحيث:

يقوم limma باستعمال نموذج إحصائي مبني على طريقة Empirical Bayes بحيث في هذه الحالة $S_0$ j و $f_0$ يمثلان تقديران للإنحراف المعياري ودرجة الحرية (Degree of freedom) تُعين من طرف النموذج و $f_{i}$ و $S_{i}$ يمثلان درجة الحرية والإنحراف المعياري للجين $i$. يمكن إستعمال هذه الطريقة مباشرة عن طريق إستعمال حزمة limma ولا داعي للدخول في تفاصيل الطريقة, المهم معرفة مبدأها, ومن أراد التفاصيل يمكنه قراءة ورقة البحث التابعة للحزمة.
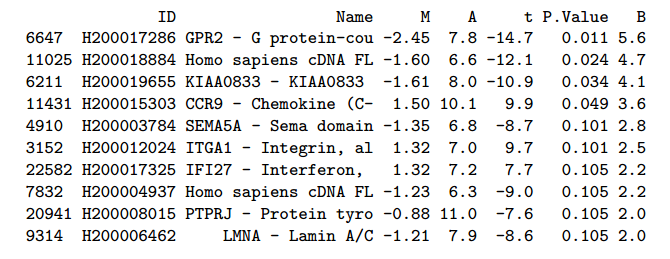
مثال عن قائمة الجينات ذات التغير الملموس بين العينتن
يجب التنبيه أيضا أنه عند الحصول على قيمة الـ p-value (والتي تمثل إحتمال أن تكون النتيجة عشوائية) يجب تصحيحها لأننا نقوم هنا بعدة إختبارات واحدة لكل جين. ويتم التصحيح باستعمال الطرق التي تتحكم في نسبة متوسط الخطأ العائلي (Familywise Error Rate أو FWER). بعد التصحيح نقوم في العادة بأخذ الجينات ذات P-value أقل أو تساوي 5% مما يعني نسبة 5% نسبة الخطأ.
8.تقسيم الجينات إلى مجموعات ودراستها:
في بعض الأحيان نريد أن نعرف ماهي الجينات التي تُعَبر بطريقة متماثلة بين العينات فنقوم بتجمعيها في مجموعات. يمكننا استخدام مجموعة من الخوارزميات لهذا الغرض, مثلا k-means, Hirachical clustering ...إلخ. بعدها يتم دراسة الوظائف الحيوية لكل مجموعة باستخدام قواميس Gene Ontology (GO) ودراسة نوع شبكات التأشير الخلوي (Signaling Pathways) التي تشارك فيها هذه الجينات بإستعمال مثلا المعلومات المتوفرة في قاعدة البيانات KEGG

مثال عن نصنيف الجينات وتحديد وظائفها (المصدر)
وعندما يتم إكتشاف دور جديد لبعض الجينات أو نقوم بشكيل بعض الفرضيات من خلال نتائج هذا التحليل نقوم بإجراء تجارب أخرى للتأكد من فرضيتنا وللتأكد من دور الجينات التي لوحظ تغيرها بشكل ملموس.
أتمنى أن يكون هذا المقال مفيدا للمهتمين بهذا المجال وأتمنى أن يتم تنبيهي لأية أخطاء في ترجمة المصطلحات أو بعض المفاهيم. وأعتذر عن طول المقال (يمكنك قرائته على مرحلتين ). في المقالات القادمة سوف أحاول شرح الجيل الجديد من التقنيات الذي يسمح لنا بقياس كمية كبيرة من الجينات وبدقة أكبر.
مصادر:
- DOV STEKEL, Microarray Bioinformatics, Cambridge University Press, 2003.
- Babu MM, Introduction to microarray data analysis. In Computational Genomics: Theory and Application. Edited by Grant RP. Norwich , Horizon Press; 2004
- Quackenbush J, Microarray data normalization and transformation, Nature Genetics , 2002

رابط المقالة : المعلوماتية الحيوية بالعربية » تقنية رقائق الحمض النووي الدقيقة (DNA Microarray) {الجزء الثاني}