السلام عليكم,
في مَقَال سَابق عرَّفنا بواحدة من اهم التقنيات المستعملة في مجال البيولوجيا و التي نتعامل مع بيانتها كثيرا في مجال المعلوماتية الحيوية والمتمثلة في تقنية الترسيب المناعي للكروماتين المتبوعة بالسَلسَلة اوما يعرف بـالـ ChIP-Seq . في هذا المقال سوف نتكلم عن الجانب العملي في تحليل بيانات ChIP-Seq وخاصة كيفية دراسة جودة البيانات المتحصل عليها.
كما شرحنا في مقال سابق, طريقة الـ ChIP-Seq هي طريقة تمزج بين الترسيب المناعي والسَلْسَلة عن طريق استعمال تقنيات الجيل الجديد, وتهدف الى تحديد أماكن ارتباط بروتين محدد عبر الجينوم. مثلا يمكننا استعامل هذه التقنية لدراسة الجينات المعدلة من طرف عامل من عوامل النسخ, مثلا ER-alpha الذي يعتبر بروتينا هاما في سرطان الثدي وبالتالي مثلا معرفة الجينات المعدلة من طرف هذا البروتين وتطوير دواء لتثبيطها او تنشيطها.
عمليا, نحتاج من مليون الى عشرة ملايين خلية و حوالي أسبوع للقيام بالعملية في المختبر, من تحظير للمكتبات وسلسلة العينات. المخطط التالي يلخص اهم المراحل في هذه التقنية (من اجل شرح مفصل, يمكن العودة الى المقال السابق):
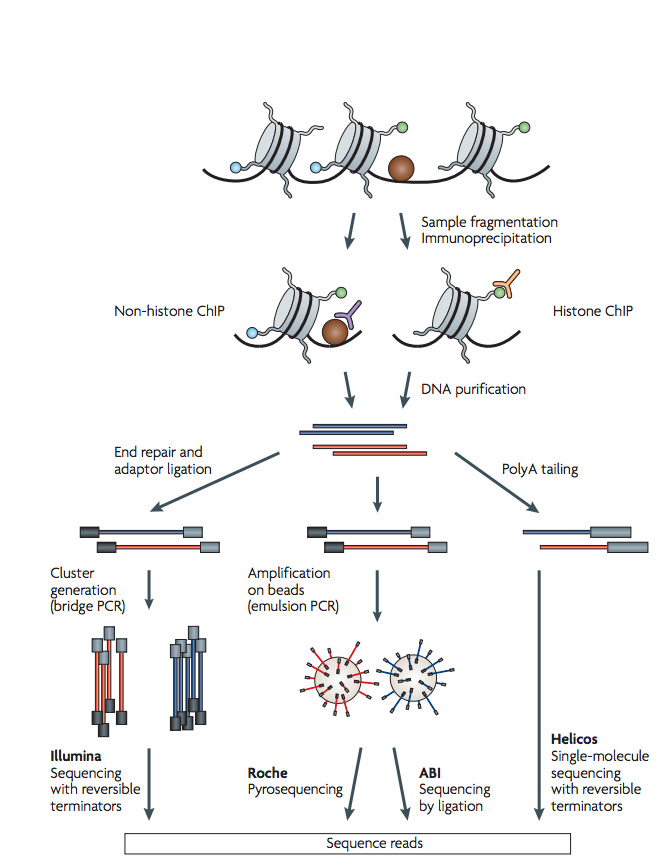
رسم بياني 1: تلخيص لمبدأ تقنية ChIP-Seq
تعتبر طريقة الـ ChIP-Seq من الطرق المهمة في مجال البيولوجيا الجزئية بحيث انها تمثل اساس العديد من التقنيات الحديثة كطريقة ChIA-PET و غيرها من الطرق. لهذا في هذا المقال سوف نستعرض بعض اهم المعايير المستعملة في تحديد جودة هذه البيانات.
مراحل تحليل بيانات ChIP-Seq
كما هو الحال في كل بيانات المعلوماتية الحيوية, يقع على عاتق الشخص المسؤول عن القيام بالتجربة ظمان جودة التجربة, لكن هذا لايعني انه سنتحصل دائما على بيانات خالية من المشاكل.
الخوض في الكلام عن كيفية تحديد جودة بيانات ChIP-Seq يقودنا الى الكلام عن اهم خطوات تحليل بيانات ChIP-Seq و المعايير التي نبحث عنها في كل مرحلة. كما لمحنا في المقال السابق تتلخص اهم مراحل تحليل بيانات الـ ChIP-Seq في الخطوات التالية:
1. تحديد جودة السلاسل: هذه الخطوة ليست محصورة في الـ ChIP-Seq فقط, بل هي الخطوة الاولى في كل انواع البيانات الناتجة عن آلات الجيل الجديد للسَلسَة. الادات المستعملة بكثرة في هذه الخطوة هي أدات FastQC. هذه الخطوة تسمح لنا بالحصول على نظرة عامة عن جودة السلاسل المتحصل عليه (اي هل تم قراءة السلاسل بشكل صحيح), ومعرفة ما اذا كانت السلاسل ملوثة ببعض السلاسل الغير جينومية كالـ Adapters المستعلمة في آلآت السلسلة. كما يمكننا الحصول على فكرة حول تاثير عملية الـ PCR على كمية السلاسل المتحصل عليها. لكن اذا كانت السلاسل ذات جودة عالية هذا لا يعني ان التجربة كانت مكللة بالنجاح. فقط تخبرنا عن جودة السلاسل الخام او الـ Raw data.
حتى وان كنا نتعحصل على معلومات حول جودة السلاسل لكن لا يمكن اعتبار هذه المرحلة من اهم المراحل في تحديد الجودة وليست متعلقة فقط ببيانات الـ ChIP-Seq.
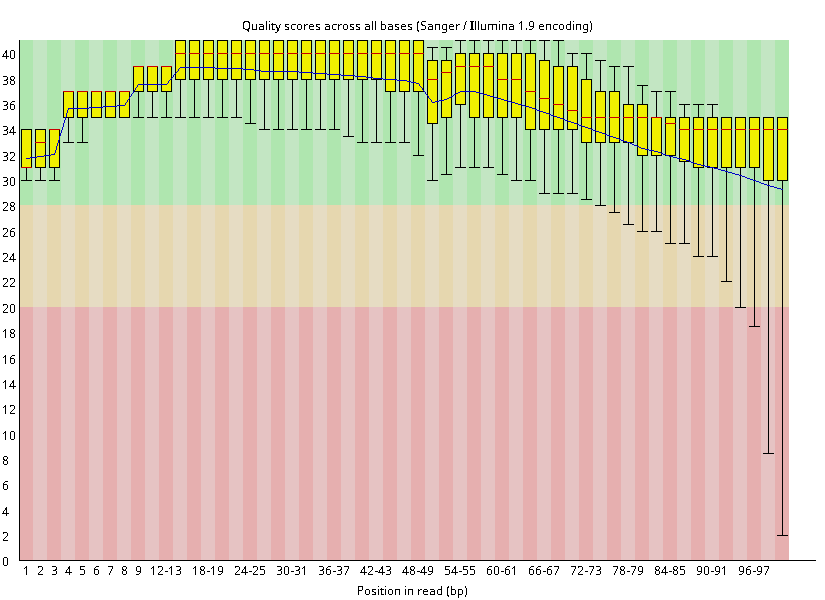 رسم بياني يوضح بيان جودة تشفير السلاسل بواسطة برنامج FastQC.
رسم بياني يوضح بيان جودة تشفير السلاسل بواسطة برنامج FastQC.
3.حذف السلاسل ذات النوعية الرديئة: اذا كان فيه مشكل في جودة البيانات, مثلا جودة الأزواج القاعدية في نهاية الـسلاسل غير جيدة او هناك بعض السلاسلة ملوثة بسلاسل الـ Adapters, يمكننا استعمال بعض الادوات مثل Cutadapt, Trim galor أو Trimmomatic لتلصليح هذه الاخطاء. في العادة, اُفَضل استعمال برنامج Trimmomatic لأن لديه خوارزميات ذكية ويمكنها تحديد بعض الاخطاء التي لم ينتبه لها المستعمل. مثلا اذا كانت لدينا سلاسل من نوع Single-end (وهي الطريقة المستعملة غالبا في الـ ChIP-Seq) يمكننا استعمال Trimmomatic كالتالي:
java -jar ./Trimmomatic SE \
-phred33 \
myChipseq.fastq \
myChipseq_filtered.fastq \
ILLUMINACLIP:${Trimmomatic_installDir}/adapters/TruSeq3-SE.fa:2:30:10:5:true \
LEADING:3 \
TRAILING:3 \
SLIDINGWINDOW:4:15 \
MINLEN:40;
2. مطابقة السلاسل للجينوم(Mapping): الآن بعدما تحَصلنا على سلاسل ذات نوعية احسن (من الاحسن استعمال FastQC مرة اخرى للتاكد من نوعية السلاسل), نقوم بمطابتها للجينوم لمعرفة مصدر هذه السلاسل. هناك عدة برامج يمكن استعمالها من اجل مطابقة السلاسل كـ Bowtie و BWA و غيرها. في هذا المثال سوف نقوم باستعمال برنامج BWA كالتالي:
echo "Mapping Single end" bwa aln -t 10 hg19.fa myChipseq_filtered.fastq > myChipseq_filtered.sai echo "Converting to sam file" bwa samp hg19.fa myChipseq_filtered.sai myChipseq_filtered.fastq > myChipseq_filtered.sam echo "converting to bam" samtools import hg19.fai myChipseq_filtered.sam myChipseq_filtered.bam echo "Sorting the bam file" samtools sort --threads 8 myChipseq_filtered.bam -o myChipseq_filtered_sorted.bam myChipseq_filtered.bam echo "indexing bam file" samtools index myChipseq_filtered_sorted.bam
4. تحديد جودة المطابقة (Mapping Quality): بعد القيام بعملية المطابقة من الاحسن التأكد من انه تم مطابقة اغلبية السلاسل للجينوم. من الادوات المستعملة بكثرة في هذه العملية هي برنامج samtools مع امري flagstat و idxstats . من بين المعايير التي ننتبه اليها: نسبة السلاسل التي تم مطابقتها, نسبة السلاسل التي تم مطابقتها لاكثر من منطقة (Multimapping).
5. تحديد مناطق الارتباط (Binding peaks calling): الآن بعدما طابقنا السلاسل الى الجينوم, يجب ان نحدد المناطق التي يتراكم فيها عدد كبير من السلاسل بطريقة غير عشوائية.لتحديد كيفية ارتباط السلاسل بطريقة عشوائية في العادة نقوم بالقيام بعملية ChIP-Seq على نفس الخلايا باستعمال مضاد حيوي غير مختص كـ IgG (في العادة لاينصح بهذه الطريقة) او استعمال الـ DNA المتحصل عليها بعد عملية تثبيت الخلايا والقيام بعملية الصوتنة (Sonication).
الهدف من استعمال عينات الفحص (Input samples) هو ان المناطق المفتوحة في الجينوم (الخالية من النكليوزمات) تكون سهلة الترسيب مقارنة بالناطق المغلوقة, مما يؤدي الى وجود مناطق غنية بالسلاسل لكن هذا لا يعني بالضرورة وجود بروتين مرتبط بتلك المنطقة. بالتالي عند استعمالنا لعينات الفحص يمكننا معرفة المناطق التي تمثل مناطق الارتباط الحقيقة من المناطق الغير حقيقية.
في العادة نستعمل برنامج macs2 من اجل تحديد مناطق ارتباط البروتين. لكن قبل استعمال هذا البرنامج يجد التأكد من الطريقة التي يرتبط بها البروتين. فاذا كان شكل القمم في مناطق الارتباط "حاد" يمكن استعمال macs2 مباشرة. اما اذا كان شكل القمم "عريضا" يمكن استعمال برنامج macs2 لكن مع استعمال امرbroad او يمكن استعمال برامج اخرى مثل Hotspot, CCAT او SICER. اما في حالة وجود خليط من الأشكال يمكن استعمال برنامج ZINBA كما هو موضح في المخطط التالي:

مخطط يوضح انواع القمم المتحصل عليها في تجارب الـ ChIP-Seq
مثلا في حالة القمم الحادة يمكن استعمال macs2 كالتالي:
macs2 callpeak -t ChIP.bam -c Control.bam -f BAM -g hs -n test -B -q 0.01
اما في حالى القمم العريضة يمكن استعماله كالتالي:
macs2 callpeak -t ChIP.bam -c Control.bam -f BAM --broad -ghs --broad-cutoff 0.1
التأكد من جودة النتائج
بعدما حددنا اماكن ارتباط البروتين و قبل الخوض في تحليل البيانات واعطاء الاستنتاجات, يجب اولا ان نتأكد من ان النتائج الاوليةالمتحصل عليها ذات نوعية جيدة. مثلا, اذا كانت هذه النتائج غير جيدة, يمكننا تغير بعض الـ parameters في برنامج macs2 او في المراحل التي قبلها, لكن في بعض الاحيان, لايوجد حل سوى اعادة التجربة البيولوجية.
في هذا المقال سوف نقوم باستعمال لغة الآر و حزمة ChIPQC لشرح المعايير المستعملة للتأكد من نوعية البيانات. كمثال سوف نقوم بتحميل ملفات من هذا الرابط وضعها في ملف chipSeq_data. هذه البيانات هي عبارة عن بيانات ChIP-Seq لعاملي النسخ ATF3 و RXL في خلايا سرطان الدم.
اذا تم تحميل الملفات بنجاح الاصل ان ترى الملفات التالية:
library("ChIPQC")
list.files("chipSeq_data/")
## [1] "BCell_Examples.RData"
## [2] "GSM847565_SL2585.gtf.gz"
## [3] "GSM847565_SL2585.table"
## [4] "GSM847566_SL2592.gtf.gz"
## [5] "GSM847566_SL2592.table"
## [6] "Homo_sapiens.GRCh37.66_chr17.gtf"
## [7] "Homo_sapiens.GRCh37.66_chr17.gtf.db"
## [8] "Homo_sapiens.GRCh37.66.gtf.gz"
## [9] "K562_ATF3_chr17_model.r"
## [10] "K562_ATF3_chr17_peaks.narrowPeak"
## [11] "K562_ATF3_chr17_peaks.xls"
## [12] "K562_ATF3_chr17_summits.bed"
## [13] "K562_RXL_chr17_model.r"
## [14] "K562_RXL_chr17_peaks.narrowPeak"
## [15] "K562_RXL_chr17_peaks.xls"
## [16] "K562_RXL_chr17_summits.bed"
## [17] "wgEncodeHaibTfbsK562Atf3V0416101AlnRep1_chr17.bam"
## [18] "wgEncodeHaibTfbsK562Atf3V0416101AlnRep1_chr17.bam.bai"
## [19] "wgEncodeHaibTfbsK562Atf3V0416101RawRep1_chr17.bigWig"
## [20] "wgEncodeHaibTfbsK562RxlchV0416101AlnRep1_chr17.bam"
## [21] "wgEncodeHaibTfbsK562RxlchV0416101AlnRep1_chr17.bam.bai"
## [22] "wgEncodeHaibTfbsK562RxlchV0416101RawRep1_chr17.bigWig"
نقوم بعدها بتحميل بيانات المطابقة bam و ملف "narrowPeak" او bed التي تحتوي على المواقع الارتباط التي تم ايجادها باستعمال برنامج macs2.
data("blacklist_hg19")
bamFile <- list.files(path = "chipSeq_data/",pattern = ".bam$",full.names = TRUE)
peakFile <- "chipSeq_data/K562_ATF3_chr17_peaks.narrowPeak"
smpls <- ChIPQCsample(bamFile[1],chromosomes = "chr17",
annotation = "hg19",
peaks = peakFile,blacklist = blacklist.hg19 )
هناك عدة معايير يمكن حسابها لاكتساب فكرة حول مدى جودة النتائج المتحصل عليها والتي يمكن تلخيصها في النقاط التالية:
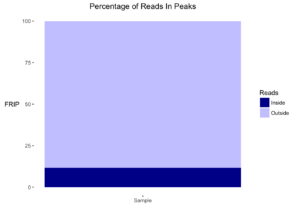
- نسبة السلاسل المتواجدة في القمم Fraction of reads in peaks (FRIP): تقوم هذه القيمة بحساب النسبة بين عدد السلاسل المتواجدة في القمم (او السلاسل المتواجدة في اماكن الارتباط) الى العدد الكلي للسلاسل (reads) المتحصل عليها في التجربية. بطبيعة الحال نريد ان تمثل اغلبية السلاسل اماكن الارتباط الحقيقية وليست فقط عبارة عن noise.
plotFrip(smpls)
- نسبة السلاسل المتموقعة في المناطق السوداء في الجينوم (Reads in black list, RiBL): عند القيام بمشروعي ENCODE و modENCODE لاحظ الباحثون وجود بعض المناطق التي تتطابق فيها السلاسل بشكل جيد مهما كان نوع التجربة وتتميز بوجود تكدس مفرط للسلاسل فيها بطريقة غير منظمة. هذه المناطق تتواجد عادة في التيلوميرات و السنترومترات. يمكن تحميل احداثيات هذه المناطق من موقع ENCODE وبعض المواقع الاخرى.
> QCmetrics(smpls)["RiBL%"] #RiBL% #0.22
- التشتت الاحصائي لتوزع عمق السَلسلة ( Dispersion of coverage): في تجارب الـ ChIP-Seq نتوقع وجود مناطق تتكدس فيها السلاسل على شكل قمم والتي تمثل مناطق الارتباط, ومناطق خالية تقريبا من السلاسل. كلما كان تكدس السلال متركز في مناطق محددة في الجينوم فهذا يدل على جودة كبيرة للتجربة. فاذا قسمنا الجينوم الى عدة مناطق متساوية الحجم وحسبا عدد السلاسل في كل منطقة, نتوقع ان تكون قيمة التشتت الاحصائي كبيرة كلما كانت جودة البيانات عالية. كما نتوقع تكون قيمة التشتت الاحصائي صغيرة في عينات الفحص.
plotCoverageHist(smpls)
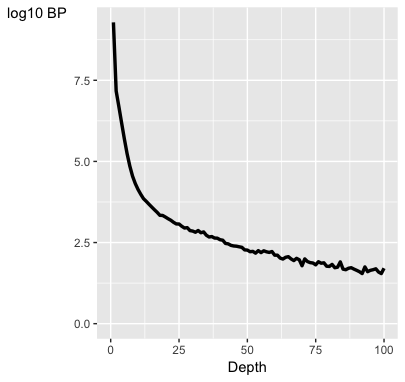
بيان يوضح نسبة تكدس السلاسل في الجينوم
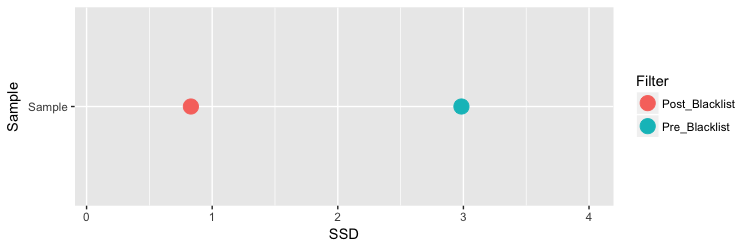
يجب توخي الحذر عند حساب نسبة توزع تكدس السلاسل, فوجود نسبة انحراف معياري كبيرة لاتعني بالظرورة ان التجربة جيدة. كما قلنا سابقا, تؤثر المناطق "السوداء" في الجينوم على نسبة توزع السلاسل في الجينوم. مثلا هنا نلاحظ ان نسبة الانحراف المعياري تقلصت كثيرا بعدما نزعنا السلاسل المتمركزة في المناطق السوداء من الجينوم.
load("chipSeq_data/BCell_Examples.RData")
plotSSD(QCsample(resExperiment,3))
- حساب التوزع المتوازي للسلاسل حول مراكز الارتباط (Cross-coverage score): بما اننا في تقنية الـ ChIP-Seq نقوم بترسيب البروتينمع سلاسل الحمض النووي التي يرتبط بها, فاننا عند قرائتها, فاننا سوف نتحصل على سلاسل (reads) من طرفي مكان ارتباط عامل النسخ, كماهو مبين في البيان التالي:
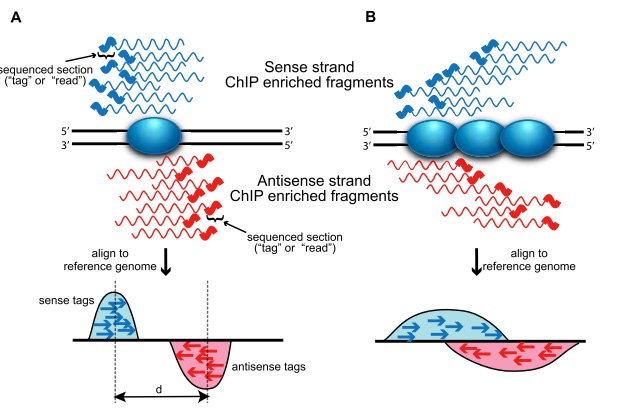
رسم تخطيطي يوضح مبدأ التوزع المتوازي لسلاسل ChIP-Seq (مصدر الصورة)
لكي نعلم موقع ارتباط البروتين, يجب ان نقوم بتوحيد القمم المتواجدة على الجهة الموجبة (ملونة بالارزق في البيان) والقمم المتواجدة على الجهة السالبة (ملونة بالاحمر). يتم هذا الدمج عن طريق ازاحة السلاسل الموجبة نحو اليمين والسالبة نحو اليسار. في حالة الاندماج التام تعطي السلاسل اقل عدد من الجزيئات القاعدية في الجينوم (نظريا العدد التي كانت تغطيه).
اذا لمعرفة نسبة هذه الازاحة نقوم بكل مرة بازاحة السلاسل بمقدار جزيء قاعدي واحد لكل من السلاسل الموجبة والسالبة ونحسب بعدها عدد الجزيات القاعدية التي تم تغطيتها. عند الازاحة بحوالي d/2 جزيء قاعدي سوف نتحصل على اقل عدد. يسمى هذا المعيار Cross-coverage score. يمكن تلخيصه في البيان التالي:
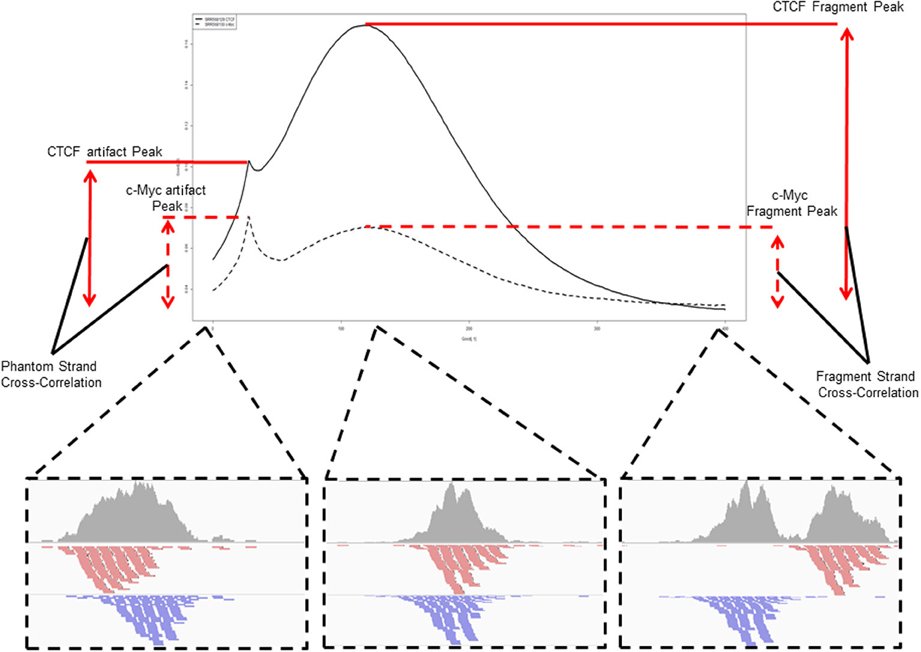
رسم تخطيطي يوضح مبدأ حساب الـ Cross-coverage score (مصدر الصورة)
تسمى اكبر قيمة بـ Fragment length (وهي قيمة متوسط حجم مكان ارتباط البروتين), كما نلاحظ وجود قمة صغيرة في المنحنى تسمى Artifact peak او Phantom peak (او القمة الشبح) لانها في الحقيقة غير دلالية وهي مساوية لحجم الـ Sequencing reads وليس لها دلالة بيولوجية.
كلما كانت النسبة  اكبر من 1 كلما دل ذلك على جودة التجربة.
اكبر من 1 كلما دل ذلك على جودة التجربة.
يمكننا حساب هذه النسبة كالتالي:
> FragmentLengthCrossCoverage(smpls) / ReadLengthCrossCoverage(smpls) #2.006214
نكتفي بهذا القدر في هذا المقال.
خلاصة
في هذا المقال لخصنا اهم مراحل تحليل و التاكد من جودة تجارب الـ ChIP-Seq. بالاضافة الى المؤشرات المذكورة هنا, هنا مؤشرات يمكننا ايضا استعمالها كحساب الـ correlation بين التجارب مثلا ونسبة التغطية حول بعض المناطق كالـ TSS وغيرها لايسع ذكرها في هذا المقال. اتمنى ان يكون هناك من استفاد من هذا المقال.
بعض المصادر:
- Elizabeth G. Wilbanks, Marc T. Facciotti (2010) Evaluation of Algorithm Performance in ChIP-Seq Peak Detection, Plos one.
- Thomas S. Carroll1 et al, Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data, frontiers in Genetics, 2014
- ChIPQC, Bioconductor,
- ChIP-Seq tutorial
- Szalkowski AM, Schmid CD (2011) Rapid innovation in chIP-seq peak-calling algorithms is outdistancing benchmarking efforts. Brief Bioinform 12:626–633.
- Jianxing Feng et al (2012) Identifying ChIP-seq enrichment using MACS, nature protocols
رابط المقالة : المعلوماتية الحيوية بالعربية » تحليل بيانات الترسيب المناعي ChIP-Seq