بسم الله الرحمن الرحيم,
من الأسئلة التي يمكن أن يتساءلها القارئ هي: "هل تتوقف قدرة آلات السلسلة عند تشفير الحمض النووي وقياس نسبة نشاط الجينات فقط؟". والاجابة هي كما هو متوقع لا . مع التقدم العلمي السريع وظهور مشاريع علمية ذات تمويل عالمي في بدايات هذا القرن مثل مشروع موسوعة عناصر الحمض النووي ( ENCODE), مشروع الميكروبيوم البشري (HMP) ومشاريع كثيرة أخرى لم تكتمل بعد اصبح استعمال آلات التسلسل من ابجديات الدراسات البيولوجية ولبنة أساسية لكثير من التقنيات.
أهمية تقنية الترسيب المناعي المقرونة بفك المتتبعات
كمثال على استعمال آلات السَلسلة , في مقالنا اليوم سوف نتطرق إلى طريقة تقنية الترسيب المناعي للكروماتين (Chromatin ImmunoPrecipitation ) المقرونة بفك المتتابعات أو اختصارا Chip-Seq . الهدف الرئيسي لهذه التقنية هو دراسة مناطق ارتباط البروتينات بالحمض النووي. كما نوهنا في المقالات السابقة هناك أنواع كثيرة من البروتينات التي ترتبط بالحمض النووي للقيام بوظائف مختلفة, فمنها من يساهم في عملية نسخ الجينات ومنها من تؤثر على بنية الكروموزم لكن كلها تساهم بطريقة أو بأخرى بالتاثير على التعبير الجيني ونشاط الخلية. يمكن تلخيص البروتينات الأكثر دراسة في الجدول التالي:
|
عوامل النسخ |
نوع من البروتينات التي ترتبط بسلاسل محددة من الحمض النووي وتساهم في تنظيم التعبير الجيني تمثل حوالي 8% من مجموع جينات الانسان |
|
الهيستون |
مجموعة من البروتينات ( H2A, H2B,H3, H4 ) توجد عادة في الخلايا حقيقية النواة يلتف حولها الحمض النووي. يشكل مركب الحمض النووي والهستون مايسمى بـ <<النكليوزوم>> . يلتف حول كل نكليوزوم 146 جزيء قاعدي. |
|
الميثيلية (Methylation) |
وهي اظافة مجموعة الميثيل إلى قاعدة السيتوزين (C) في الحمض النووي فتتحول إلى 5-ميثيل سايتوزن عن طريق انزيم DNMT. تحدث هذه الاضافة في السلاسل CG (تسمى عادة CpG) . في العادة الجينات التي تحتوي على مجموعة الميثيل تكون صامتة أما الجينات التي لاتحتوي تكون نشطة. تسمى المناطق الغنية بسلاسل الـ CpG ب <<جزر الـ CpG>> في العادة تكون هذه الجزر قريبة من المحفزات (Promoters) و المحسنات (Enhancers) الجينية. |
|
تغيرات الهيسيتون |
مجموعة من التفاعلات الكيميائية تحدت في أذيال بروتينات الهيستون تؤدي إلى تغيير بنية النكليوزوم فينفتح أو ينغلق. انفتاح النكليوزم يسمح بالرتباط البروتينات بالحمض النووي وانعلاقه يعيق هذا الارتباط. يمكن للهيستونات ان تتعرض للمجموعة من التغيرات كالمَثْيَلة, الأَسْتَلة, الفسفرة,...إلخ لكل منها تأثير على التعبير الجيني وتكون في العادة في أماكن محددة من الحمض النووي |
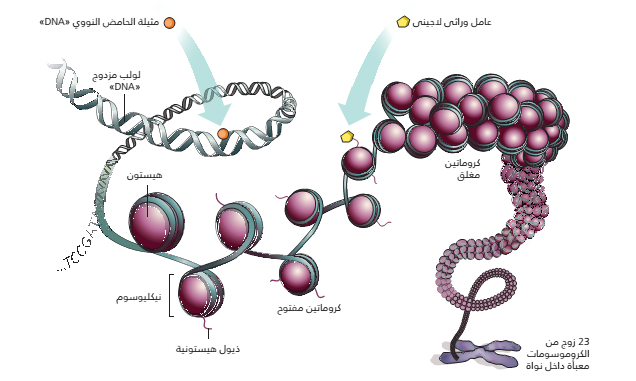
رسم تخطيطي يبين مبداء التغيرات الاجينية (المصدر)
اكتشف العلماء أن الكثير من الاختلافات النمطية والسلوكية عند الكائنات الحية ومجموعة كبيرة من الأمراض ليست نابعة من اختلاف في سلسلة الحمض النووي لكنها ناتجة عن تغيرات لاجينية (مَيْثلة الحمض النووي, تغيرات الهستون). فمثلا في بعض الأمراض يتم إسكات أو تنشيط مجموعة من الجينات عن طريق مَيْثلة مُحفِز أو مُحَسن جين من الجينات مما يؤدي إلى اعاقة وصول عوامل النسخ.
ادى ظهور تقنيات السلسلة وبالتالي تقنية الترسيب المناعي المرفوق بفك المتتابعات (Chip-Seq) الى اعادة سطوع نجم علم الوراثة اللاجينية (Epigenetics) خاصة بعد انتهاء مشروع ENCODE وانتاج اكثر من 1600 مجموعة بيانات لأكثر من 100 نوع من الخلايا ورسم خريطة ارتباط عوامل النسخ والتغيرات الاجينية.
اذن فدراسة اماكن ارتباط البروتينات بالحمض النووي يعطينا عدة معلومات عن كيفية عمل عوامل النسخ والجينات المعدلة من طرف هذه العوامل كما تمكننا أيضا من معرفة الفروق الاجينية بين الخلايا وفهم نشوء الامراض. السؤال المطروح الآن هو ماهو مبدأ عمل تقنية الـ Chip-Seq؟
مبدأ تقنية الترسيب المناعي:
تعتمد تقنية الترسيب المناعي على مبدا بسيط وهو "بما أن البروتين مرتبط بالحمض النووي, فاذا تمكننا من أخذ هذا الجزء وسلسلته ثم مطابقته للجينوم يمكننا معرفة تواجد هذا البروتين". السؤال الآن هو "كيف نقطع ذالك الجزء من الجينوم؟", يمكننا فعل ذالك في ثلاث خطوات:
1. في الخطوة الأولى نقوم باستعمال محلول الفورمالدهيد (formaldehyde ) لتثبيت حالة الخلية (لأنها في حالة دينامكية) وبالتالي تثبت البروتينات المرتبطة بالحمض النووي,
2. الخطوة الثانية: بعدها نستعمل طريقة الصوتنة (sonication) والتي تتمثل في استعمال قوة الموجات الصوتية للتقطيع الحمض النووي بطريقة عشوائية.
3. الخطوة الثالثة: نستعمل مضادات حيوية التي يمكنها التعرف بشكل محدد على البروتين الذي نريد دراسته فتلتصق هذه المضادات الحيوية بالبروتين وتقوم بترسيب قطع الـحمض النووي التي تحتوي على البروتين الذي نريد دراسته.
بعد الحصول على قطع الحمض النووي نقوم ننزع البروتين والشوائب الاخرى ثم نقوم بسلسلة هذه القطع, نتحصل بعدها على ملف FASTQ يحتوي على صيغة هذه السلاسل (في العادة يكون طول السلاسل مابين 25 إلى 35 جزيء قاعدي). لمعرفت موقع هذه السلاسل نقوم بمطابقتها للجينوم المرجعي باستعمال برامج مثل Bowtie أو BWA أو غيرها من برامج المطابقة.

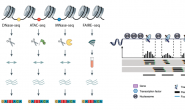
مخطط يوضح مبدأ طريقة Chip-Seq (الصورة الأصلية)
اكتشاف أماكن الارتباط
بما أن البروتين يرتبط فقط باماكن محددة في الجينوم فهذا بعني أن عملية مطابقة السلاسل سوف تؤدي إلى الحصول على مناطق غنية بالسلاسل (تكون على شكل قمم) ومناطق خالية أو فقيرة من السلاسل. الهدف اذا هو تصميم بعض الخوارزميات التي تقوم بتحديد القمم التي تمثل مناطق ارتباط بروتين حقيقية و المناطق التي تمثل ضجيج أو أخطاء في السَلْسَلة.

مخطط يوضح مبدأ المطابقة وتحديد القمم (المصدر)
بما أن بعض السلاسل المتحصل عليها يتطابق مع السلسلة الموجبة للحمض النووي (البيان الممثل باللون الأزرق) وجزء يتطابق مع السلسلة السالبة (البيان الممثل باللون الأحمر) تقوم الخوارزميات بتوحيد هذان المنحنيان في منحنى واحد في العادة يقوم باتخاذ المركز بين القمتين كمركزللقمة الجديدة. بعدها تقوم الخوارزمية بتحديد القمم ذات الدلالة الاحصائية, تختلف طريقة تحديد القمم ذات الدلالة الاحصائة من خوازمية إلى اخرى, فبعض الخوارزميات تقوم بتوليد سلاسل موزعة بطريقة عشوائية موزعة على حسب توزيع Poisson وبعدها تقارن الشبه بينها وبين توزيع السلاسل المتحصل عليها في عملية Chip-Seq. أما الطريقة الثانية فتتطلب القيام بتجربة Chip-Seq أخرى لكن هذه المرة باستعمال مضاد حيوي عام ثم نقارن بين توزيع السلاسل في هذه العينة (عينة الضبظ) والعينة الرئيسية وبعدها تحديد القمم ذات الدلالة الاحصائية بحساب قيمة P-value.
تمثل كل قمة وضعية في الجينوم وتكون محددة برقم الكروموزم (الصبغي) ومكان بدايتها ونهايتها و قيمة الـ P-value التي تحدد مدى ثقة الخوارزمية في صحة هذه القمة (نذكر أنه كلما كانت قيمة الـ P-value صغيرة كلما زادت الدلالة الاحصائية). بطبيعة الحال تختلف النتائج باختلاف النتائج لكن في العادة تكون القمم ذات الدلالة الاحصائية العالية مشتركة بين كل الخوارزميات. لهذا ينصح باستعمال برنامج تحديد الققم واحد لدراسة كل العينات وذالك لتفادي الوقوع في أخطاء أثناء المقارنة.
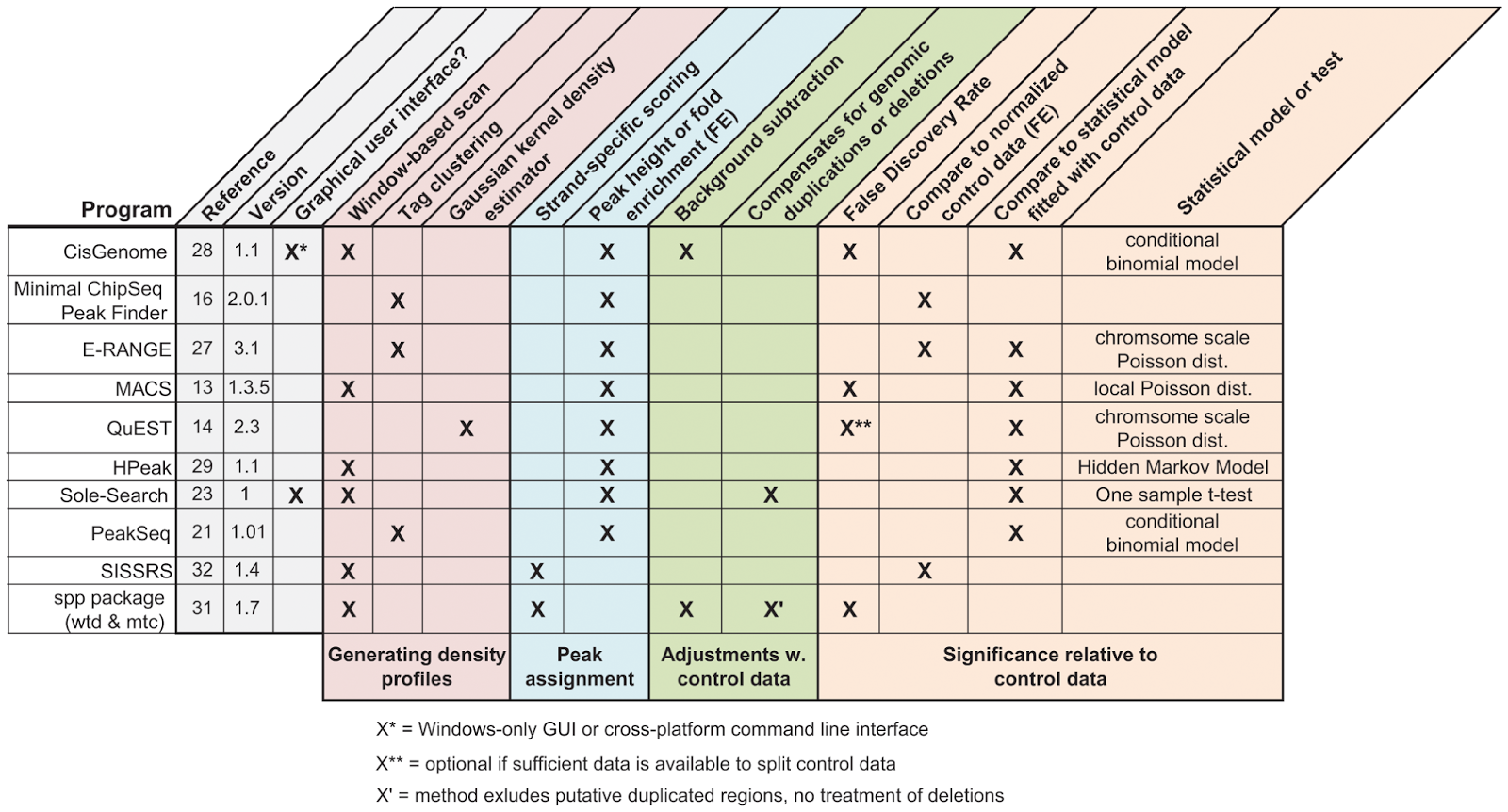
جدول يقارن بين خصائص برامج تحديد القيم الأكثر استعمالا(المصدر)
استخدامات تقنية الـ Chip-Seq
السؤال المطروح الآن هو : " حسنا الآن تحصلت على قائمة المواقع التي يرتبط بها البروتين ماذا يمكنني أن أستنتج منها؟", بطبيعة الحال الجواب مرتبط بأهداف الدراسة التي تريد القيام بها, لكن هناك مجموعة من خطوات تحليل البيانات الكلاسيكية (ملخصة في البيان ) التي تمكنك من الحصول على فكرة ابتدائية على كيفية عمل البروتين والأماكن التي يرتبط بها.

كخطوة ابتدائية يمكنك القيام بتحاليل استكشافية (Exploratory analysis) مثلا يكننا إستعمال متصفح جينوم مثل UCSC أو IGV أو غيرها والمقارنة بالاستعمال العين المجردة بين نتائج العينات المختلفة (مثلا خلايا سليمة وخلايا سرطانية) وملاحظة الاختلاف. للحصول على تحليل أكثر دقة يمكن أن نحليل نوعية الاماكن التي يرتبط بها البروتين في العينات المختلفة, مثلا ما هي نسبة أماكن الارتباط القريبة من المحسنات وماهي نسبة الأماكن القريبة من المحفزات وهل يرتبط البروتين بالأكسونات أو الانترونات,...إلخ. يكمننا أيضا دراسة التباين في شكل الرتباط بين مختلف العينات مثلا يمكن دراسة الاماكن المشتركة والاماكن المختلفة وهل هناك اختلاف كبير أم لا بين العينتين وأن كان فيه اختلاف ماهو الفرق.
يمكن أيضا استعمال بعض خوارزميات تصنيف البيانات ونقوم بتجميع أماكن الارتباط التي المتشابهة مع بعض (مثلا أماكن الارتباط القوية والضعيفة, أو تصنبفيها على حسب موقها في بنية الجين,... إلخ)
من بين أهم التحاليل أيضا هو دراسة صيغة السلسلة التي يرتبط بها البروتين (motif analysis), يعتمد مبدأ هذا التحليل على مقارنة صيغة السلاسل التي يرتبط بها البروتين ومحاولة ايجاد الصيغ المتكررة في هذه السلاسل. يمكننا استعمال هذه المعلومة لمعرفة هل يرتبط هذا البروتين بصيغة محددة في الحمض النووي وماهي البروتينات التي تمتلك صيغات متشابهة وهل هناك تنافس بين هذه البروتينات للارتباط بتلك الصيغة.
بمقارنة الصيغ (motifs) المختلفة مع قاعدة بيانات الصيغ يمكننا معرفة البروتينات التي ترتبط بجوار هذا البروتين وبالتالي انشاء نظرية حول العلاقة بينهما ومدى تأثيرهما على التعبير الجيني. توجد عدة برامج لتحليل الموتيفات مثل برنامج MEME-Chip و Homer . في العادة تمثل صيغة السلسلة التي يرتبط بها البروتين (أو الموتيفات) على شكل بيان logo, يمثل حجم كل حرف في هذا البيان احتمال ارتباط البروتين بالجزيء القاعدي (الصورة). يمكن أيضا كتابة هذه النتائج على شكل مصفوفة أوزان المواضع (Position weight matrix)
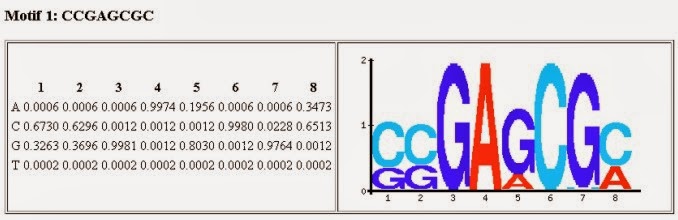
صورة توضح مبداء بيان اللوغو لmotif يتكون من 8 جزيئات قاعدية (المصدر)
من التحاليل التي يمكن القيام بها ايضا هو دراسة علاقة أماكن أرتباط البروتين بالتعبير الجيني. مثلا يمكننا القيام بتجربة RNA-Seq في مختلف العينات والقيام ببعض العمليات الاحصايئة لاختبار فرضية وجود علاقة بين ارتباط البروتين والتعبير الجيني. يمكننا ايضا اسعمال طرق تحليل مجموعة الجينات (Gene set analysis) باستعمال الـ Gene ontology لمعرفة المجموعات الوضيفية التي تنتمي إليها هذا الجينات.
وهناك أيضا عدة طرق لتمثيل البيانات ودراستها لكن هنا قدمنا فقط لمحة شاملة بعض الشيء عن هذه التقنية وبعض التحليلات الاكثر استعمالا. ربما في المقالات القادمة سوف نتطرق بطريقة مفصلة لكيفية تحليل هذه البيانات.
وفي الأخير أتمنى أن يكون هذا المقال مفيدا.
اذا كانت فيه أخطاء في ترجمة بعض المصطلحات العلمية أرجوا التنبيه
مصادر:
- Bardet AF, He Q, Zeitlinger J, Stark A: A computational pipeline for comparative ChIP-seq analyses. Nat Protoc 2012, 7:45-61.
- Park P. J. (2009). ChIP–seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 10.1038/nrg2641
- T. S. Furey, ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions, Nature Reviews Genetics, vol. 13, no. 12, pp. 840–852, 2012.
رابط المقالة : المعلوماتية الحيوية بالعربية » دراسة أماكن ارتباط البروتينات بالحمض النووي باستعمال طريقة Chip-Seq