بسم الله الرحمن الرحيم,
على الرغم من وجود أدوات تساعدنا في التعامل مع مختلف ملفات السَلسَلة, كملفات .fastq التي تحتوي على صيغة السلاسل أو ملفات .bam أو .sam المتحصل عليها بعد القيام بعملية المطابقة مع الجينوم المرجعي, إلا أن هذه الأدوات في بعض الأحيان لا تعطيك المرونة الكافية للقيام ببعض العمليات أو للحصول على بعض الاحصائيات. لهذا يضطر الباحثون إلى كتابة بعض السكريبتات للقيام بذلك.
يمكن استعمال عدة لغات للتعامل مع ملفات FASQ أو bam الـ BED أو الصيغ الأخرى ويعود أمر اختيار لغة البرمجة التي تريد استعمالها لاختيارك الشخصي. من بين اللغات الأكثر استعمالا نجد لغة الـ Pearl حيث يمكنك مثلا استعمال حزمة Bioperl التي تحتوي على عدة دوال تسمح لك بقراءة مختلف أنواع الملفات, إلأ ان Pearl لا يعطيك المرونة الكافية للتعامل مع المصفوفات أو انشاء بعض الرسومات البيانية وتكون السكريبتات في العادة طويلة. لهذا السبب نجد أن كثيرا من المبرمجين يتجهون نحو Python أو لغة الـ R. لكن لغة الآر تبقي من الحلول المفضلة, نظرا لجمعها بين السهولة وبساطة السكرييتات ولتوفر العديد من حزم المعلوماتية الحيوية.
في هذا المقال سوف نعطي بعض الامثلة بلغة الآر عن العمليات الأكثر استعمالا كالمطابقة بين السلاسل والقيام بعمليات تقاطع بين الاحداثيات الجينومية والتعامل مع الملفات التي تحتوي على نتائج السَلسَلة كـ BAM و SAM.
حزمة Biostrings
عند تحميل لغة الآر تأتي مزودة ببعض الدوال للتعامل مع السلاسل الحرفية (Strings) العادية دون الأخذ بعين الاعتبار هل هي سلسلة بيولوجية أم فقط سلسلة حرفية عادية كما هو الحال في كافة اللغات الأخرى. فباستعمال دوال لغة الآر الأصلية يمكنك القيام بكل العمليات المعروفة من قلب و قص و بحث على جملة توافق قالبا من القوالب عن طريق استعمال التعابير القياسية (Regular expressions), ... إلخ.
لكن عند الوصول إلى التعامل مع السَلاسل الجينومية سوف تلاحظ أن هذه الدوال لا تكفي وحتى وان كانت كافية فستلاحظ انها غير ملائمة في حالة البيانات الكبيرة. يعود هذا إلى عدة أسباب من بينها :
- ان الخوارزميات مصممة للتعامل مع جمل ذات ابجديات كبيرة (26 حرف + الارقام + الرموز الخاصة) بينما تتكون السلال البيولوجية من ابجدية بسيطة 4 حروف بالنسبة للـ DNA أو الـ RNA و 20 حرفا بالنسبة للبروتين بالاضافة لحرف N الذي يدل ان الصيغة غير محددة.
- عملية مطابقة السلاسل البيولوجية تعتمد على مبدا مختلف عن عمليات المطابقة في السلاسل الحرفية, حيث أنها تأخذ بعين الاعتبار مبدا الادراج (Insertion) و الحذف (Deletion) وعدد الحروف الغير متطابقة ( mismatch count),... إلخ. لهذا تم تصميم خوارزميات لهذا الغرض كخوارزمية سميث و واترمان المشهورة.
أساسيات التعامل مع حزمة Biostrings
في لغة الآر يمكن استعمال حزمة Biostrings التي توفر بيئة متطورة تسمح بالتعامل مع السلاسل حيث تتوفر على العديد من الخوازميات السريعة و الكائنات لحواية السلاسل بطريقة ذكية لاستعمال الذاكرة وغيرها من المرافق، للتعامل السريع مع مجموعة كبيرة من السلاسل البيولوجية.
تٌعَرِف حزمة Biostrings خمسة فئات (Classes) أساسية:
| XString | وهي الفئة القاعدية التي ترث منها كل الفئات الأخرى وهي فئة افتراضية ولا يمكن انشاء كائنات منها. |
| BString | فئة فرعية تسمح بالتعامل مع كل أنواع السلاسل ( البيولوجية أو السلاسل العادية) |
| DNAString | فئة فرعية للتعامل مع سلاسل الحمض النووي |
| RNAString | فئة فرعية للتعامل مع سلاسل الحمض النووي الريبوزومي |
| AAString | فئة فرعية للتعامل مع السلاسل البروتينية ( سلاسل الأحماض الأمينية) |
يمكن انشاء كائنات من هذه الفئات كالتالي:
يتم التعامل مع هذه الكائنات وكانك تتعامل مع جدول حروف حيث يمكن حساب الطول وقراءة الحروف باعطاء فقط رقم الحرف كما هو مبين في المثال:
في حالة إذا ما أردت التعامل مع مجموعة من السلاسل وليس سِلسلة واحدة فقط يمكنك استعمال الكائنات : DNAStringSet , RNAStringSet , AAStringSet و BStringSet. من بين مزايا هذه الكائنات هو انها تمكنك من القيام بنفس العملية على مجموعة من السلاسل مرة واحدة دون كتابة كود معقد. مثلا في بعض الحالات تكوم نوعية سَلْسَلة الجزيئات القاعدية في مؤخرة كل سلسلة رديئة (phred-score < 20) فنقوم بحذف مثلا العشر جزيئات القاعدية الأخيرة من كل سلسلة باستعمال أمرsubseq.
كما يمكنك انشاء سلاسل فرعية من سلسلة ما دون اللجوء إلى عمل نسخ جديدة وذلك عن طريق استعمال النوافذ (Views), لهذه الخاصية أهمية كبيرة خاصة إذا كنت تتعامل مع سلسلة طويلة كسلسلة كروموزم من الكروموزومات.
في المثال التالي سوف نستعمل الكروموزوم 1 من الجينوم البشري (BSgenome.Hsapiens.UCSC.hg19) المتوفرة في حزمة BSgenome (اظافتا إلى جينومات الالكائنات النموذجية ( model organisms) ) وننشى نوافذ حول بعض الجينات التي نفرض أننا مهتمين بها.
بعدها يمكننا مثلا القيام بعمليات لحساب نسبة متتابعات الـ GC أو القيام بعملية الـ motif analysis لايجاد المتتابعات المتكررة.
واحدة من أهم الدوال التي توفرها حزمة Biostrings هي دوال مطابقة السلاسل نلخص بعضها في الجدول التالي:
| matchPattern , countPattern | لإيجاد مواضع تطابق سلسلة ما في سلسلة أخرى أو حساب عدد المطابقات (*v). |
| vmatchPattern, vcountPattern | إيجاد مواضع تطابقة سلسلة ما في مجموعة من السلاسل أو حساب عدد المطابقات في كل سلسلة (*v). |
| pairwiseAlignment | مطابقة سلسلتين باستعمال خوازمية Needleman-Wunsch للمطابقة الكلية و خوارزمية Smith-Waterman للمطابقة الجزئية |
| matchPWM , countPWM | المطابقة باستعمال مصفوفة أوزان (Pair-wise Weight Matrix) محددة من طرف المستعمل |
| matchPDict , countPDict | لإيجاد مواضع تطابق مجموعة من السلاسل في سلسلة أخرى أو حساب عدد المطابقات (*v). |
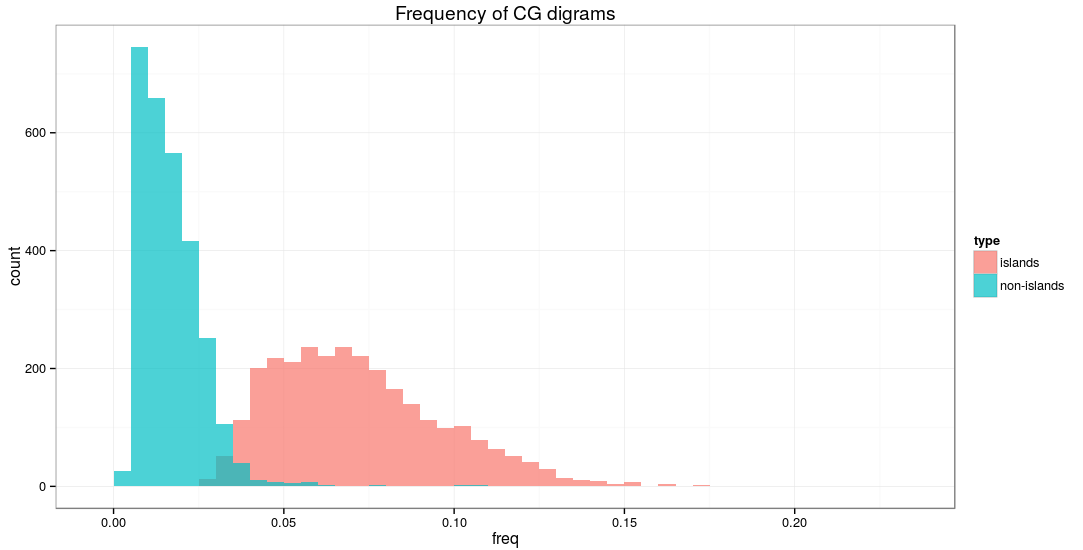
في المثال التالي سوف نستعمل مثال مقتبس من هذا الدرس, حيث نقوم باستعمال دالة vcountPattern لحساب نسبة تواجد سلاسل CG في مجموعتين من المناطق في الجينوم رقم 8. في العادة تتكتل سلاسل الـ CG بشكل غير عشوائي في مناطق تسمى جزر الـ GpC و التي تتواجد في العادة من القرب محفزات الجينات مما يجعلها ذات أهمية وظيفية.
من المخطط البياني نلاحظ أن نبسة الـ CG في الجزر أكبر بشكل ملحوظ من نظيرتها في المناطق الأخرى.
نكتفي في هذا المقال بهذا الجزء وسوف نحاول التعريف بحزم ودوال اخرى للتعامل مع السلاسل كدوال التعامل مع المجالات الجينومية و قراءة ملفات مثل FASTQ و BAM.
رابط المقالة : المعلوماتية الحيوية بالعربية » التعامل مع السلاسل البيولوجية بلغة الآر(الجزء الأول)