بسم الله الرحمن الرحيم
مع إنتهاء مشروع الجينوم البشري ظهرت هناك رغبة كبيرة للحصول على أكبر قدر من سلاسل الحمض النووي من عدة أناس و عدة كائنات ومعرفة مقدار تشابة وإختلاف هذه السلاسل. رأينا أن تقنية سانغر كانت هي المسيطر لمدة عقدين من الزمن في مجال تشفير الحمض النووي, لكن تظافرت الجهود من أجل تطوير تقنيات تدمج بين خاصية السرعة والسعر الرخيص وأدت بالفعل إلى ظهور مايسمى بالجيل الجديد لتقنيات السلسلة (Next Generation Sequencing Technologies) وأصبحت الآلات القائمة على تقنية سانغر تسمى بآلات الجيل الأول (First Generation Sequencing).
مع نهاية مشروع الجينوم البشري، قامت شركة 454 بإطلاق اول آلة سلسلة سنة 2005، بعدها بسنة قامت شركة Solexa بإطلاق آلة Genome Analyzer لتليها شركة Agentcourt بآلتها SOLiD. ولاقت هذه الآلآت رواجا حيث أصبحت من الآلات الكلاسيكية في كبريات المخابر فور ظهورها. وفي اقل من سنة من قامت شركات أخرى بشراء الشركات الأم، حيث اشترت شركة Roche شركة 454 واشترت Applied Biosystems شركة Agentcourt واشترت illumina شركة Solexa
إن ظهور تقنيات الجيل الجديد من الآت السلسلة أدى إلى إحداث تغيرات جذرية في مناهج البحث العلمي المتبعة قديما و غيرت بشكل ملموس طريقة نظر العلماء للمسائل البيولوجية وكيفية التعامل معها. حيث مر العلماء من فترة إستعمال تقنيات السلسلة لتشفير سلاسل الحمض النووي و الجينات فقط إلى عهد تستعمل فيه تقنيات السلسلة لدراسة أغلبية المسائل البيولوجية, فبإظافة تغيرات بسيطة يمكننا دراسة البنية ثلاثية الأبعاد للكرموزوم كما يمكننا دراسة المناطق المفتوحة والمغلقة من الحمض النووي, ودراسة أنواع الـ RNA التي تترجم إلى بروتين ومراكز إرتباط عناصر النسخ, وغيرها من الأشياء التي لايمكن حصرها في هذه الأسطر مما أدى إلى جعل هذه التقنيات من أبجديات البحث البيولوجي حيث لايمكن الإستغناء عنها.
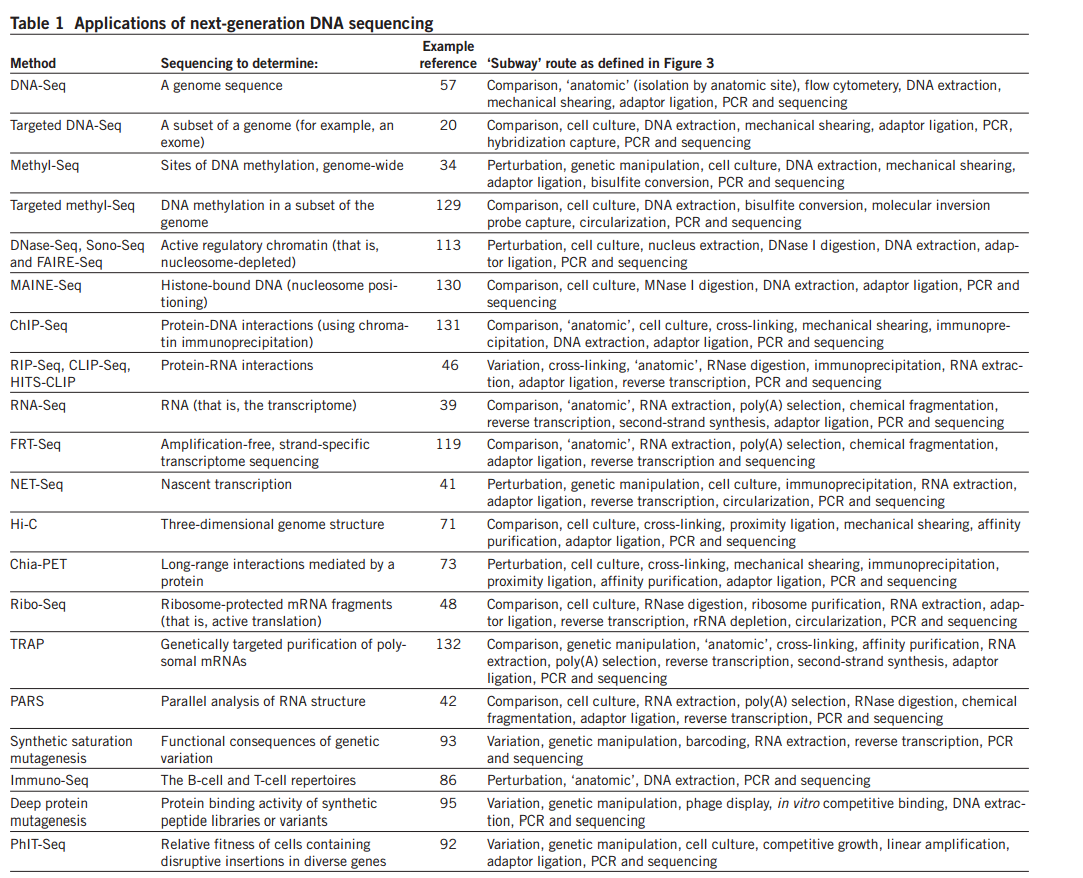
قائمة ببعض التقنيات التي تعتمد على آلآت الجيل الجديد (إضغظ على الصورم من اجل التكبير) (المصدر)
في هذا المقال سوف نحاول ان نعطي صورة كاملة بعض الشيئ عن هذه التقنيات مع توضيح مبدأ عمل كل واحدة منها. سوف نقوم بالتركيز على الآلات الأكثر استعمالا في السوق حيث سنقدم آلات Roche/454, illumia/Solexa و Helicos Bioscience.
تكمن نقاط اختلاف هذه الآلات في أربعة نقاط وهي: تحظير النموذج، طريقة التشفير، طريقة معالجة الصور و طريقة معالجة البيانات
1- تحضير النموذج ( Template Preparation ):
يقصد بالنموذج هنا سلسلة من الحمض النووي تتكون من جزئين، جزئ نعرف صيغته، مثلا adaptor sequence يمكن أن تلتحم مع primer عام ، و الجزئ الثاني يتمثل في سلسلة غير معروفة الصيغة وهي التي نريد أن نسلسلها. ولأن هذه التفنيات تعتمد على مبدا إضافة جزيئات قاعدية مشعة يجب مضاعة كمية الحمض النووي المستخرجة للحصول على كمية إشعاع جيد.
تعتمد أغلب الآلآت مبدا من المبدأين التاليين:
أ- التضخيم بإستعمال المستعمرات (Clonally amplified templates) :
تعتمد هذه الطريقة على مبدا تضخيم (أو نسخ) أجزاء الحمض النووي التي تم الحصول عن طريق التقطيع العشوائي للحمض النووي بدون اللجوء إلى وسط خلوي كالبكتريا مثلا. تتم هذه العملية بإستعمال طريقة emulsion-based clonal amplification أو إختصارا emPCR وهي طريقة مبنية على نفس مبدا طريقة تفاعل البلمرة التسلسلي PCR (وهي طريقة كلاسيكية تتستعمل في المخابر من أجل صنع نسخ للسلاسل). ففي البداية يتم إظافة سلسلة مؤلِف (Adaptor) إلى نهاية كل قطعة من قطع الحمض النووي وذالك لتسهيل تظخيمها باستعمال بوادئ PCR او PRC Primer. بعدها يتم إلصاق كل جزء من هذه الأجزاء بالكريات بطريقة تمكننا على الحصول على سلسلة واحدة في كل كرية ( أنظر الصورة) وبعدها توضع كل كرية (Bead) في قطرة ماء وتوضع هذه الكريات في محلول زيتي وبهذا تكون كل كرية مفصولة عن الآخرى. وفي المرحلة الأخيرة يتم القيام بعملية النسخ باستعمال طريقة PCR (زيادة و خفض الحرارة لفك الروابط بين السلسلتين واستعمال أنزيم DNA Polymerase من أجل القيام بالنسخ), بعد نهاية العملية تتكون على ظهر كل كرة ملايين من النسخ للسلسة الملتصقة وبذالك يمكننا الحصول على عدد كبير من النسخ. في العادة يكون طول كل السلسلة الواحدة في كل كرية من 200 إلى 250 جزئ قاعدي.

صورة توضح مبدأ تحضير النماذج في آلات 454 (المصدر)
أما آلآت illumina فتعتمد على مبدأ تضخيم آخر حيث أنها تسعمل صفيحة دعم صلبة تحتوي على بوادي أمامية (Forward Primers) و بوادئ خلفية (Backward Primers) ويتم بعد ذالك لحم كل سلسلة بالبوادئ.في البداية تكون السلال بعيدة عن بعضها البعض بحيث تشكل مجموعات (Clusters) منفصلة بعد التضخيم. تسمى هذه العملية بطريقة التضخيم الجسري (Bridge templating) حيث تقوم عملية التضخيم على رفع درجة الحرارة فتنحني كل سلسلة وتلتصق بالبادئ المقابل مشكلةً بذالك جسرا, في هذه المرحلة يتم نسخ السلسلة إنطلاقا من البادي الأقرب, بعدها يتم خفض درجة الحرارة فتستقيم السلاسل مرة أخرى, وبتكرار العملية عدة مرات تتشكل لدينا مجموعات منفصلة على سطح الصفيحة حيث تمثل كل مجموعة نسخة لسلسلة واحدة. يمكننا الحصول في صفيحة واحدة على حوالي مليون مجموعة. في العادة يكون طول كل السلسلة الواحدة من 100 إلى 150 جزئ قاعدي. 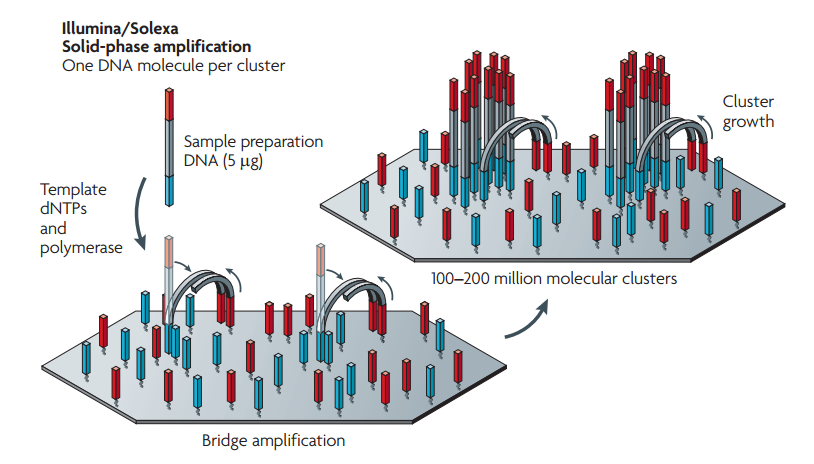
رسم توضيحي لمبدأ Bridge templating المستعمل من طرف illumina (المصدر)
ب- التضخيم باستعمال جزبئ واحد (Single Molecule templates):
على الرغم من أن طريقة استعمال التضخيم باستعمال المستعمرات تقوم بالتضخيم بشكل جيد إلا أنها ليست سهلة التحضير وتتطلب كمية كبيرة من الـ DNA مابين $20 \mu g$ و $30 \mu g$ لهذا تم تطوير طريقة التضخيم باستعمال جزي واحد والتي تحتاج إلى أقل من $1 \mu g$ . كما تتميز هذه الطريقة بعدم حاجتها لإستعمال PCR. في هذه الطريقة يتم تبيت كل جزبئ إلى بادئ (Primer) على سطح الصفيحة وتكون السلاسل إما متقاربة أو متباعدة وهذه الطريقة متبعة من طرف آلآت Helicos BioSciences.

رسم توضيحي للمبدأ المستعمل من طرف آلآت Helicos, سلاسل متقاربة (يمين) وسلاسل متباعدة (يسار)(المصدر)
هناك طريقة أخرى متبعة من طرف شركة Pacific Biosciences حيث أنها عوض أن تثيت السلسلة بالصفيحة وبعدها إستعمال أنزيم DNA Polymerase للقيام بقراءة النسخ, تقوم التقنية بثبيت الانزيم على الصفيحة وإظافة السلال المزودة بالبوادئ, وتسمح لنا هذه الطريقة باستعمال سلاسل طويلة تصل إلى 14kb.

رسم توضحي للمبدأ المستعمل من طرف شركة Pacific Biosciences (المصدر)
2- السلسلة ومعالجة الصور (Sequencing and Imaging):
في المرحلة يتم تشفير السلاسل التي تم تحضيرها في المرحلة السابقة وكما يمكن استنتاجه أن الطريقة تختلف من آلة إلى أخرى لكن كلها تعتمد على مبدأ إضافة جزيئات قاعدية مشعة و تحليل الصور لمعرفة نوع الجزيئ القاعدي الموجود في السلسلة. ا- آلآت Roche 454: بعد تحضير الكريات يتم وضع كل كرية في صحيفة تحتوي على ثقوب (تسمى أيضا آبار) بحيث يتسع الثقب الواحد لكرية واحدة فقط. تعتمد طريقة الترميز في هذه الآلآت على مبدأ سلسلة البايرو (المطورة من طرف مصطفى روناجي) والتي يتم خلالها إستعمال جزيئة البايرو فوسفات التي تشع بعد قيام الـ DNA polymerase بلحم الجزيئات القاعدية. باختصار يقوم مبدأ عمل التقنية كاللآتي, يتم وضع الكريات المحضرة سابقا مع مجموعة من الكريات الصغيرة جدا تحتوي على مكونات كيميائة تسهل عملية الإشعاع, وفي كل مرة يتم إضافة كمية من مكون الـ dNTP ويتم في كل مرة قراءة نسبة الضوء بعد حدوث عملية التهجين (Synthesis) وبالتالي تحديد نوع الجزيئ القاعدي. بطبيعة الحال هناك أخطاء في القراءة حيث أن نسبة التقدم في كل سلسلة في الكرية قد لا تكون متساوية مما يؤي إلى التشويش على نسبة الإشعاع.

رسم بياني يوضح مبدأ التهجين في آلآت Roche 454 (المصدر)
ب-آلآت illumina: تقوم آلآت Illumina على مبدأ أستعمال طرق دورية (Cyclic Methods) حيث تتكون من ثلاث مراحل وهي إظافة الجزيئ القاعدي, أخذ الصورة و القطع. في المرحلة الأولى يتم إظافة الجزيئات القاعدية A,C,G,T مشعة و معدلة بطريقة تسمح باظافة جزء واحد فقط عند القيام بعملية التهجين, لما يتم تهجين هذا الجزئ القاعدي يقوم باطلاق ضوء (لدينا أربعة ألوان) والذي يتم إلتقاط صورة له, بعد إلتقاط الصورة يتم غسل الصفيحة و نزع القاعدة الكيمائية المضافة إلى الجزيئ القاعدي من أجل السماح باستمرار عملية التهجين في الدورة القادمة.

رسم بياني يوضح مبدا السلسلة في آلآت Illumina (المصدر)
ج- آلات Helicos: تقوم آلآت Helicos على نفس مبدأ عمل آلآت Illumina إلا أن تستعمل لونا واحدا ويتم إظافة جزيئ قاعدي فقط في كل مرة عكس Illumia التي تضيف كل الجزيئات القاعدية لكن كل واحد بلون خاص, يمكن تلخيص التقنية في الرسم البياني التالي:
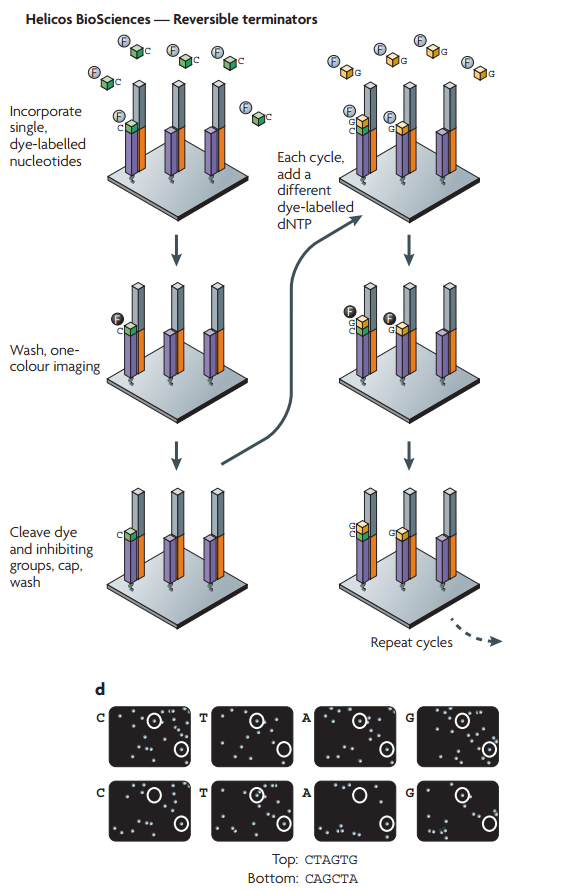
رسم بياني يوضح مبدا السلسلة في آلآت Helicos (المصدر)
د- آلآت Pacific Biosciences: تمثل هذه الآلآت آلات مابعد الجيل الجديد لآلآت السلسلة إذ أنها تسمح لنا بالسلسلة بطريقة مباشرة وبدون اللجوء إلى حيلة إيقاف عملية التهجين بعد إضافة كل جزيئ قاعدي. على الرغم من أن استعمال هذه الآلآت لازال ضعيفا في السوق إلا أنها سوف تسيطر على سوق آلآت السلسلة في المستقبل القريب.
في هذه التقنية يتم تثبيت أنزيم DNA plymeraze في ثقوب بحيث تكون سلسلة واحدة ملتصقة بالأنزيم في كل ثقب ويتم إظافة جزيئات قاعدية تشع عند حدوث عملية التهجين ويتم قياس قيمة الإشارة المولدة كما هو موضح في الشكل.
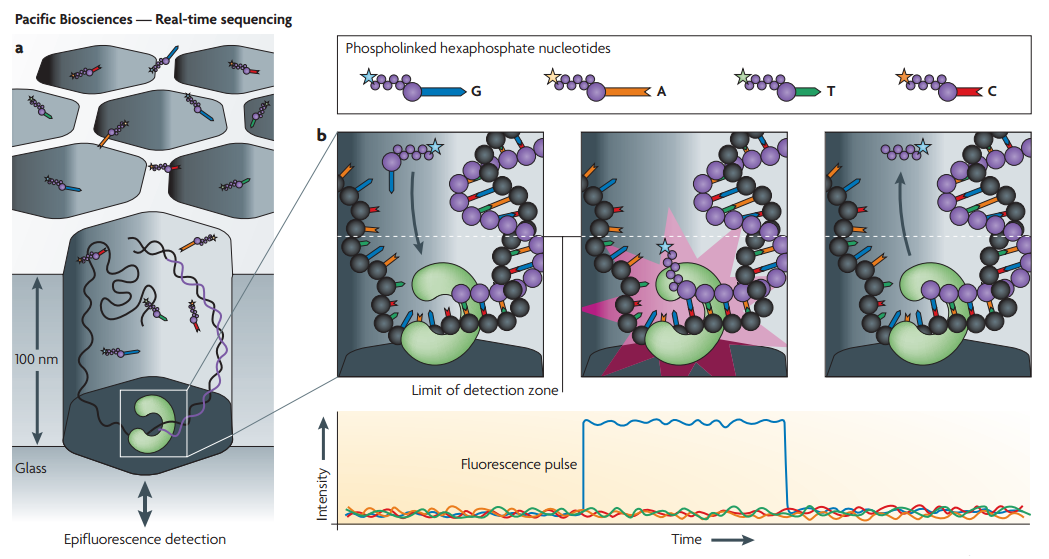
رسم بياني يوضح مبدا السلسلة في آلآت Pacific Biosciences (المصدر)
3-مطابقة السلاسل للجينوم (Read Mapping):
في حالة توفر ضغة الجينوم, مثلا الجينوم البشري, فتم مطابقة السلاسل المتحصل عليها للجينوم البشري ومعرفة مكان قدوم هذه السلاسل. فمثلا يمكننا أخذ سلاسل الـ RNA الموجودة في خلية سراطانية وسلسلتها ثم نقوم بمطابقتها للجينوم البشري ومعرفة الجينات المتأثر بالسرطان.
لكن هناك بعض المشاكل التي تواجهنا في هذه المرحلة, حيث لايمكننا تحديد مكان السلسلة في الجينوم لأن الجينوم يحتوي على مناطق متشابهة كما أن السلاسل المرمزة في العادة تكون قصيرة حوالي 100 جزئ قاعدي. لكن آلآت Pacific Bioscience تسمح لنا بالحصول على سلاسل أكبر وبالتالي إمكانية المطابقة بطريقة أدق.
من بين الأدوات الأكثر إستعمالا في المطابقة يوجد برنامج Bowtie, BWA,MAQ, TopHat,... إلخ
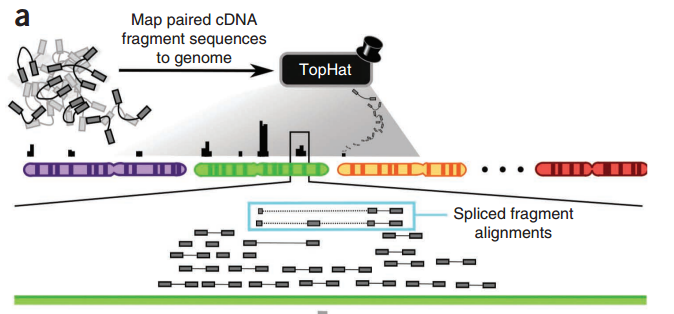
رسم توضيحي لمبدأ مطابقة السلاسل بالستعمال برنامج TopHat (المصدر)
في حالة عدم معرفة صيغة الجينوم الأصلي نقوم بعملية التجميع من البداية(de-novo Assembly) حيث يتم تجميع السلاسل التي تحتوي على أجزاء متشابهة معا للحصول على صيغة الجينوم الأصلي مثل لعبة تجميع الصور المقطعة.
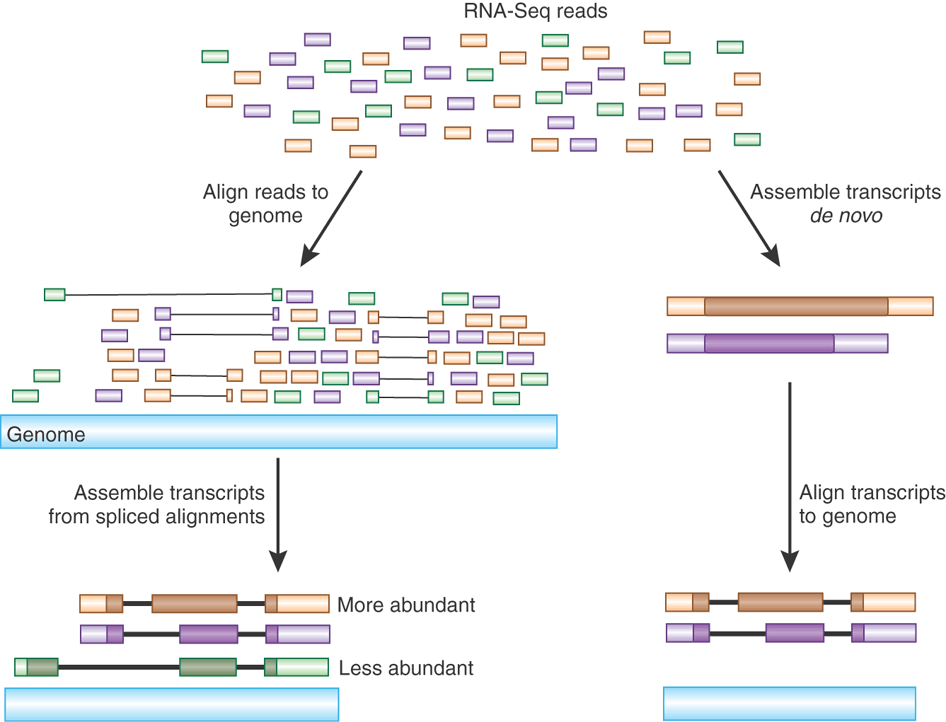
تلخيص لمبدئي مطابقة وتجميع السلاسل (المصدر)
وفي الخلاصة أتمنى أن يكون هذا المقال مفيدا رغم الإختصار في بعض المناطق, وأرجوا التنبيه لأية أخطاء. في المقالات القادمة ان شاء الله سوف نتكلم عن كيفية تحليل البيانات وسوف نقدم بعض التقنيات مثل RNA-Seq, Chip-Seq, ChIA-PET, ...إلخ كل في مقال.
المصادر:
- Michael L. Metzker, Sequencing technologies — the next generation, Nature Reviews Genetics 11, 31-46 (January 2010)
- Cole Trapnell et al, Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation, Nature Biotechnology 28, 511–515 (2010)
- Brian J. H. and Michael C. Z., Advancing RNA-Seq analysis, Nature Biotechnology 28, 421–423 (2010)
- Yongjun Chu and David R. Corey, RNA Sequencing: Platform Selection, Experimental Design, and Data Interpretation, Nucleic Acid Ther. Aug 2012; 22(4): 271–274
رابط المقالة : المعلوماتية الحيوية بالعربية » نظرة على الجيل الجديد لآلآت السلسَلة