
بسم الله الرحمن الرحيم,
في المقال السابق قدمنا فكرة عامة عن لغة الـ R لكن لم نقدم مثالا يوضح كيفية استعمالها من أجل تحليل البيانات الحيوية. في هذا المقال سوف نحاول أن نأخذ مثالا عمليا نعطي من خلاله نبذة عن تحليل البيانات باستعمال R.
البيانات المستعملة:
في هذا المقال سوف نأخذ كمثال البيانات المستعملة من طرف الباحث Sabina Chiaretti وزملائه من أجل دراسة خصائص التعبير الجيني في مرض ابيضاض الدم اللمفاوي الحاد (Acute Lymphocytic Leukemia) أو اختصارا ALL وهو نوع من أنواع سرطان الدم ( Leukemia) والذي يتميز بالتكاثر الغير طبيعي للخلايا الأرومية الليمفاوية (lymphoblasts) في نخاع العظم وهي عبارة عن خلايا دم بدائية غير ناضجة وغير متمايزة. يصيب هذا المرض كلا من الخلايا البائية (B-Cells) و التائية ( T-Cells ).

رسم تخطيطي يوضح الفرق بين التركيبة الخلوية للدم في الحالة العادية وفي حالة السرطان (مصدر الصورة)
في هذه الدراسة قام الباحثون بقياس التعبير الجيني لـ 12625 جين باستعمال رقائق الحمض النووي ( Microarray ) لـ 128 مريض شخصوا حديثا بمرض الـ ALL حيث أن مجموعة منهم مصابة في الخلايا البائية و مجموعة في الخلايا التائية.
تحميل البيانات وتحضير البيئة البرمجية:
سبب اختيارنا لهذا المثال هو توفر البيانات مباشرة في موقع Biocondctor في حزمة ALL بالاضافة إلى هذه البيانات نحتاج أيضا إلى تثبيت الحزم التالية affy و RColorBrewer. يمكننا تثبيت هذه الحزم باستعمال الأوامر التالية:
source("http://bioconductor.org/biocLite.R")
biocLite("affy")
biocLite("ALL")
## RColorBrewer is located in the CRAN
install.packages("RColorBrewer")
library(affy)
library(ALL)
## load the data
data(ALL)
اخذ نظرة على البيانات:
لو تكلمنا بمصطلحات لغة البرمجة, نلاحظ أن البيانات محفوضة في كائن ( object ) من نوع ExpressionSet المصمم خصيصا لحفض بيانات التعبير الجيني لتجارب الرقائق الدقيقة حيث أنه يحتوي على نتائج كل عينة و معلومات عامة عن التجربة كإسم التقنية المستعملة, عدد الجينات, معلومات حول المرضى والورقة البحثية المنشورة... إلخ. يمكننا رؤية بعض هذه المعلومات كالتالي:
> ALL ExpressionSet (storageMode: lockedEnvironment) assayData: 12625 features, 128 samples element names: exprs protocolData: none phenoData sampleNames: 01005 01010 ... LAL4 (128 total) varLabels: cod diagnosis ... date last seen (21 total) varMetadata: labelDescription featureData: none experimentData: use 'experimentData(object)' pubMedIds: 1468442
مثلا لو أردنا معرفة نوع البيانات المتوفرة عن المرضى يمكننا استعمال دالتي phenoData و varMetadata كالتالي:
> pd <- phenoData(ALL)
varMetadata(pd)
labelDescription
cod Patient ID
diagnosis Date of diagnosis
sex Gender of the patient
age Age of the patient at entry
BT does the patient have B-cell or T-cell ALL
remission Complete remission(CR), refractory(REF) or NA. Derived from CR
CR Original remisson data
date.cr Date complete remission if achieved
t(4;11) did the patient have t(4;11) translocation. Derived from citog
t(9;22) did the patient have t(9;22) translocation. Derived from citog
cyto.normal Was cytogenetic test normal? Derived from citog
citog original citogenetics data, deletions or t(4;11), t(9;22) status
mol.biol molecular biology
fusion protein which of p190, p210 or p190/210 for bcr/able
mdr multi-drug resistant
kinet ploidy: either diploid or hyperd.
ccr Continuous complete remission? Derived from f.u
relapse Relapse? Derived from f.u
transplant did the patient receive a bone marrow transplant? Derived from f.u
f.u follow up data available
date last seen date patient was last seen
هناك أمران يستعملان بكثرة لمعرفة كيفية توزيع البيانات, الأول وهو table وتسعمل في حالة التعامل مع البيانات الفئوية (مثلا: ذكر, أنثى أو 1,2,3) وتسمح باحصاء عدد البيانات من كل صنف. الثانية هي hist لرسم الجدول التكراري للبيانات وتستعمل في حالة البيانات المستمرة أو المتقطعة. في العادة ترسم المخططات التكرارية مع دالة الكثافة الاحتمالية باستعمال الأمر density.
يمكننا استعمال هذه الأوامر للتعرف على توزيع البيانات التي لدينا , مثلا كم عدد المرضي المصابين في الخلايا التائية والخلايا البائية, التوزيع العمري للمرضي, ... الخ.
> BT <- table(ALL$BT) > BT B B1 B2 B3 B4 T T1 T2 T3 T4 5 19 36 23 12 5 1 15 10 2 ## Total number of B patients > sum(BT[1:5]) [1] 95 ## Total number of T patients > sum(BT[6:10]) [1] 33 number of male and female patients > table(ALL$sex) F M 42 83 > hist(ALL$age,xlab="Age",main="Age distribution")
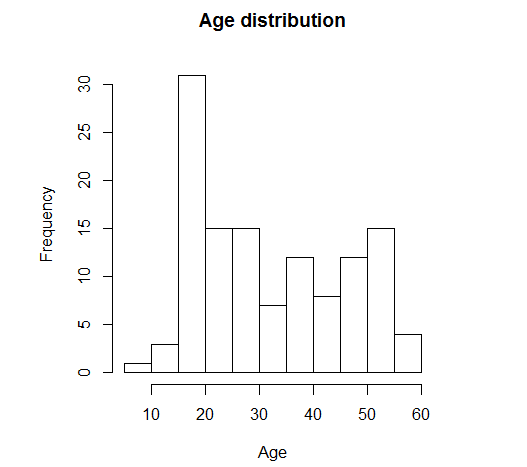
مخطط تكراري لتوزيع الفئات العمرية
نلاحظ من المخطط التكراري وجود قمتين واحدة كبيرة متمركزة قرب عمر 18 سنة والثانية أصغر قرب 52 سنة مما يعطينا فكرة أولية على أن هناك العينات تحتوي على نسبة كبيرة من الشباب ونسبة اخرى من الكهول.
دراسة تعبير بعض الجينات:
لتبسيط المثال نفرض أنه لدينا الجينات التالية المعروفة في مساهتها في موت الخلية (apoptosis) ونريد أن نرى الفرق بين نسبة نشاطها في المرضى المصابين في الخلايا التائية و المرضى المصابين في الخلايا البائية. حيث نعلم أن الخلايا السرطانية هي خلايا تنموا بطريقة غير طبيعة ولا تستجيب إلى الإشارات القادمة من المحيط الخلوي التي تدعوها عن التوقف عن التكاثر أو الموت. كما أنها لا تستجيب إلى الأدوية الكيمائية التي تحفزها على الموت. لنفرض إذا أننا ندرس الجينات التالية:
> genes [1] "ABL1" "Tial1" "SIVA1" "foxo3b" "FOXO3" [6] "CDKN1A" "LOC100271831" "MAPK3" "ABL1" "BCLAF1" [11] "LOC731605" "Tie1" "DAP" "ABL1" "CUL1"
بما أننا نتسعمل الرقائق الدقيقية يجب علينا أولا أن نحول أسماء الجينات إلى الـ ID المقابل في الرقائق الدقيقة, يمكننا استعمال R او مباشرة موقع DAVID فتحصل على الـ ID التالية:
>gn.list <- c("36199_at","39020_at","2031_s_at", "39723_at",
+ "1635_at","1636_g_at", "39730_at","34740_at",
+ "41763_g_at","38050_at","1000_at","1001_at")
مثلا نأخد التعبير الجيني لهذه الجينات في 15 مريضا مصابا في الخلايا البائية و 15 مريضا مصاب في الخلايا التائية كالتالي:
> gn <- featureNames(ALL) > t <- is.element(gn,gn.list) > small.eset <- exprs(ALL)[t,c(81:110)])
من بين المخططات التي يمكننا استعمالها لأخذ نظرة أولية عن نشاط هذه الجينات في هذه المجموعة من المرضى هي الخرائط الحرارية, حيث يمكننا رسم مصفوفة كل سطر فيها يمثل جين وكل عمود يمثل مريضا من المرضى. ونلون كل خانة على حسب نسبة نشاط الجين. للحصول على معلومات اكثر، نُرتب المرضى في المصفوفة على حسب نسبة تشابه التعبير الجيني. يمكننا القيام بهذا اتوماتيكيا باستعمال خوارزمية من خوارزميات التجميع (clustering) مثلا هنا نستعمل الـ hierarchical clustering.
قبل رسم الخريطة الحرارية نختار مجموعة من الألوان ذات دلالة لكي نتمكن من الرؤية بوضوح كالتالي:
> library(RColorBrewer) > hmcol <- colorRampPalette(brewer.pal(10,"RdBu"))(256)
ثم نعيد تسمية المرضى حيث نرمز بـ T للمرضى المصابين في الخلايا التائية و B للمرضى المصابين في الخلايا البائية.
> cell <- c(rep('B',15),rep('T',15))
> colnames(small.eset) <- cell
بما أن ترتيب المرضى والجينات سوف يتغير في المصفوفة بعد القيام بالتجميع يمكننا اضافة عمودان ملونان على حاشية المصفوفة. في العمود الخاص بالمرضى نلون بالاحمر المرضى المصابين في الخلايا التائية وبالأزرق المرضى في الخلايا البائية. بالنسبة للجينات نعلم ان كل الجينات لها دور في موت الخلية ماعدا الجين الأول و الثاني لذا سنولونهما بالأحمر والباقي بالأزرق.
> csc <- rep(hmcol[50],ncol(small.eset)) > csc[cell=='B'] <- hmcol[200]
> gn.apop <- c(0,0,rep(1,10)) > rsc <- rep(hmcol[50],nrow(small.eset)) > rsc[gn.apop==1] <- hmcol[200]
الآن يمكننا رسم الخريطة الحرارية :
> heatmap(small.eset,scale="column",col=hmcol, ColSideColors=csc,RowSideColors=rsc)
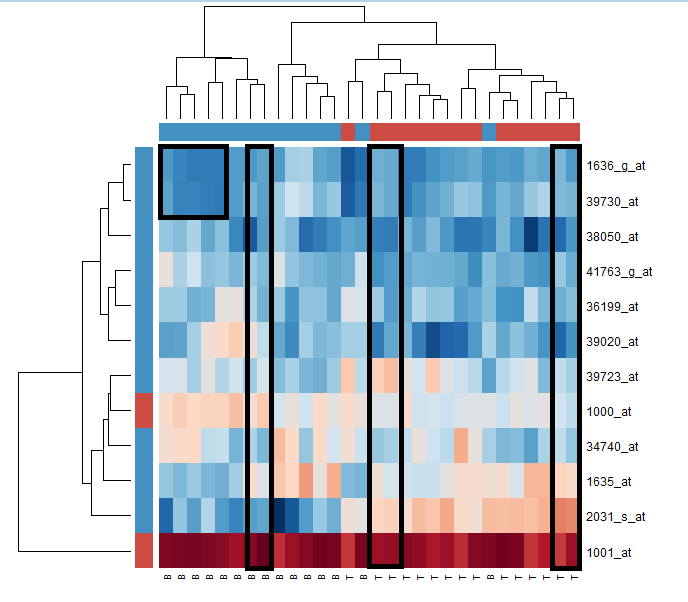
خريطة حرارية تبين تعبير الجينات المختارة في الـ30 مريض
نلاحظ أن بعض المرضى (اعمدة) لهم تعبير جينى متقارب في أغلبية الجينات و بعضهم يتشابه في بعض الجينات (موضحة في المربع الأسود) لكن هذا المخطط بهذه الطريقة لا يعطينا معلومات كافية أو ربما ليس من السهل قراءته من الوهلة الأولى . يمكننا أن نبذل جهد و نعتمد على نتائج خوارزمية التجميع الموضحة على حواشي البيان حيث تم تجميع المرضى على شكل مخطط هرمي لكن يمكننا التمثيل بطريقة أحسن.
سيكون من الاحسن إذن لو استطعنا حساب مدى التشابه بين التعبير الجيني بين المرضى عن طريق استعمال طريقة حسابية تعطينا قيمة تمثل هذا التشابه. لو اعتبرنا كل عمود على أنه شعاع, يمكننا حساب المسافة بين هذه الأشعة باستعمال طرق كلاسكية مثلا بحساب المسافة الاقليدية (Euclidian distanc) والتي تحسب المسافة بين شعاعين $X$ و $Y$ كالتالي:
$d=\sqrt{\sum_{i=1}^{n} (x_i-y_i)^2}$
يمكن أيضا حساب معامل الارتباط ( في العادة تكون النتائج أحسن). من بين معاملات الارتباط المستعملة بكثرة هو معامل سبيرمان ( Spearman correlation coefficient) كالتالي:
$\rho = \frac{\sum_i(x_i-\bar{x})(y_i-\bar{y})}{\sqrt{\sum_i (x_i-\bar{x})^2 \sum_i(y_i-\bar{y})^2}}$
يمكننا كتابة دالة لحساب هذه المعاملات أو مباشرة استعمال الدالة cor.dist لحساب معامل ارتباط سبيرمان و الدالة euc لحساب المسافة الاقليدية. هذه الدوال متواجدة في حزمة bioDist في موقع Bioconducor.
في المثال التالي سوف نقوم بحساب قيم تشابه التعبير الجيني للجينات في كل المرضى (أو بصيغة أخرى التشابه بين أسطر الخريطة الحرارية في المثال الأول) باستعمال كلا الطريقتين (معامل الارتباط والمسافة) و حساب تشابه التعبير الجينى بين المرضي في كل الجينات (الأعمدة). وذللك كالتالي:
> biocLite("bioDist")
> d.gene.cor <- cor.dist(small.eset)
> heatmap(as.matrix(d.gene.cor),sym=TRUE,col=hmcol,
+ main='Between-gene correlation (Pearson)',
+ xlab='probe set id',labCol=NA)
> d.gene.euc <- euc(small.eset)
> heatmap(as.matrix(d.gene.euc),sym=TRUE,col=hmcol,
+ main='distances Between-gene (Euclidean)',
+ xlab='probe set id',labCol=NA)
> d.sample.cor <- cor.dist(t(small.eset))
> heatmap(as.matrix(d.sample.cor),sym=TRUE,col=hmcol,
+ main='Between-sample correlation (Pearson)',
+ xlab='cell type')
> d.sample.euc <- euc(t(small.eset))
> heatmap(as.matrix(d.sample.euc),sym=TRUE,col=hmcol,
+main='distances Between-sample(Euclidean)',
+ xlab='cell type')
يمكننا تجميع هذه الخرائط الحرارية في صورة لنتحصل على نظرة أشمل
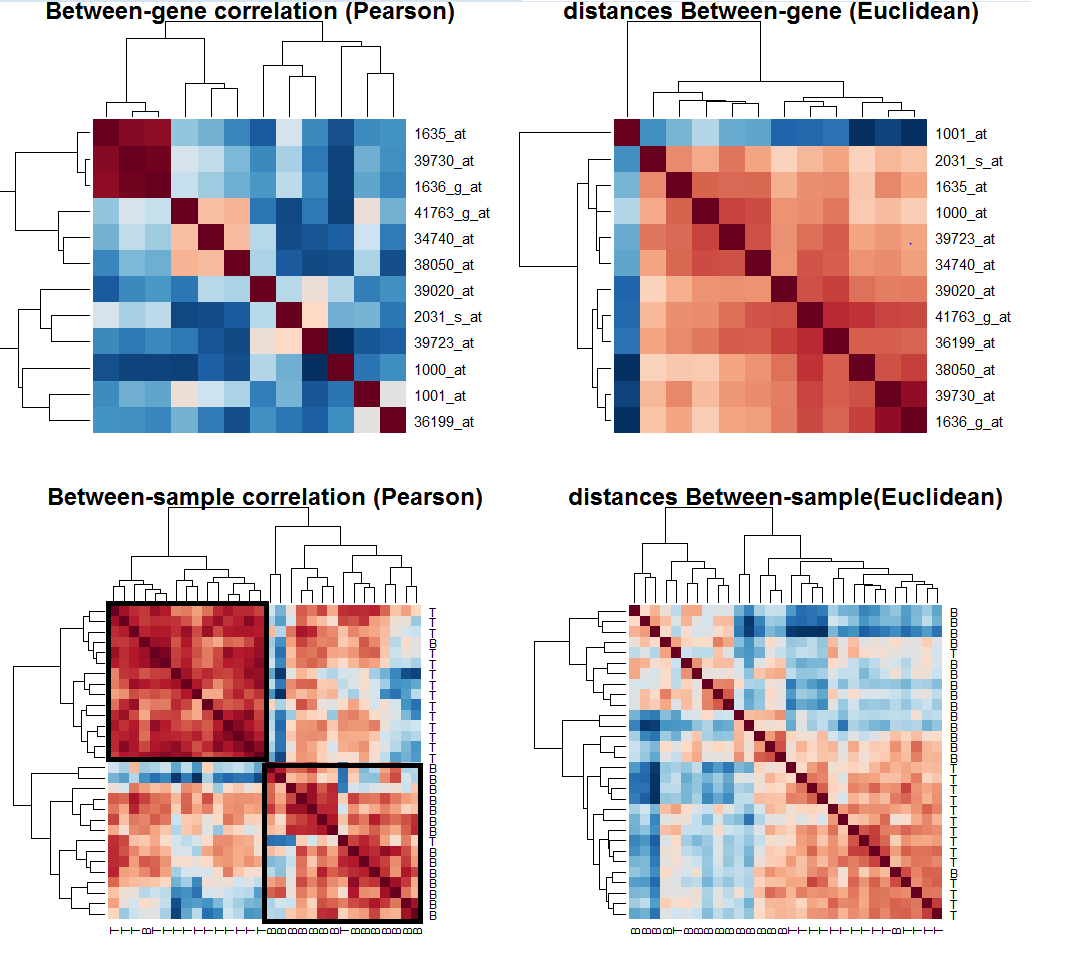
مخطط حراري يمثل نسبة تشابه التعبير الجيني في المرضى أو بين الجينات باستعمال طرق مختلفة
نلاحظ أن استعمال معامل الارتباط لحساب التشابة بين الجينات وبين المرضى يعطينا نظرة أحسن. فمثلا نلاحظ أنه يكننا نقسيم المرضى إلى مجموعتين (الممثلة بالمربعين بالأسود) الأولى على اليمين تحتوي على المرضى في الخلايا البائية لكن يتخللهم مريض في الخلايا التائية. في المربع الذي على اليسار نلاحظ تجمع للمرضى في الخلايا التائية يتخللهم مريض مصاب في الخلايا البائية.
يمكننا أن تستنتج أن التعبير الجينى يتغير على حسب الخلايا المريضة, أما بالنسبة للمريضين الذان لم يصنفا من أقرانهما هناك احتمالان إما انه كانت هناك بعض الاخطاء في التجربة أو ربما حقا النتيجة صحيحة. مثلا يمكن للباحث ان يأخذ هذان العينتان ويقوم بدراسة ادق لفهم سبب تشابهما مع الصنف الآخر أو القيام بالتجربة مرة أخرى بالنسبة لهذين المريضين والتأكد.
نأمل أن هذا القدر من الأمثلة سوف يساعد القارئ على الحصول نوعا ما على فكرة عن كيفية استعمال لغة الـ R للتحليل البيانات حتى و ان لم يكن المثال شاملا (مثلا كيف تختار الجينات, ماهي الادوات الاحصائية التي يمكن استعمالها, ... الخ).
هذا المثال مقتبس من هذا الدرس.
رابط المقالة : المعلوماتية الحيوية بالعربية » مثال عن استعمال لغة الآر لتحليل البيانات الحيوية







{kind=link}