بسم الله الرحمن الرحيم
رأينا في المقال السابق أن الجيل الجديد من آلآت السلسلة فتح آفاقا جديدة في مجال الأحياء وغَير طريقة التعاطي مع المسائل البيولوجية على الرغم من عدم مرور عشر سنوات على ظهور هذه التقنيات. أُولى تطبيقات هذه التكنولوجيا كانت تهدف إلى دراسة مجموعة النواسخ (Transcriptome) أو بعبارة أخرى كل مايتم نسخه من الحمض النووي من جينات وغيرها من أنواع الـ RNA الغير الناسخة للبروتين (التي لا تترجم إلى بروتينات مثل ال micro-RNA ) واكثشاف الهيئات المتساوية (Isoforms) وهي معلومات لم يكن من السهل الحصول عليها قبل ظهور هذه التقنيات.
إن دراسة وتشخيص مجموعة النواسخ (Transcriptome) تسمح لنا بفهم وتحديد العناصر الوظيفية في الخلية واكتشاف الجزيئات التي تساهم في تشكيل الخلايا والأنسجة مما يساعدنا في فهم تطور الأمراض والأورام وكيفية مكافحتها. تعتبر تقنية الـ RNA-Seq امتدادا طبيعا لتطبيقات آلآت الجيل الجديد للسَلْسَلَة حيث أصبح الآن بمقدورنا دراسة مجموعة النواسخ (Transcriptome) على نطاق الجينوم وعلى مستويات متعددة.
مبدأ تقنية الـ RNA-Seq :
يمكننا تلخيص مبدأ تقنية الـ RNA-Seq في الخطوات التالية( لاحظ المخطط):
1- تحظير مجموعة من الـ RNA من خلية أو مجموعة خلايا من نفس النوع وانشاء سلاسل cDNA تكميلية.
2-إنشاء مكتبة EST باضافة ملائِمات (Adaptors) لنهايات السلاسل التكميلية
3- سَلْسلة هذه السلاسل باستعمال آلآت الجيل الجديد للسَلْسَلة.
4- اذا كنا نعرف مسبقا اللصيغة الكاملة للجينوم المستعمل ( مثلا الانسان حيث تم تشفيره في مشروع الجينوم البشري), يمككننا مطابقة سلاسل الـ RNA التي تحصلنل عليها مع الجينوم المرجعي وبالتالي نتمكن من معرفة الجينات النشطة.
تعمد تقنية الـ RNA-Seq على حساب عدد السلاسل التي تطابق جزء من الجينوم (مثلا: جين) وبالتالي فالمناطق التي تطابق عدد كبير من السلاسل تكون أكثر نشاطا. في العادة يكون طول السلاسل من 30 إلى 400 جزيء قاعي على حسب الآلة المستعملة فالآلآت التي تشفر السلاسل قصيرة تسمح لنا بمعرفة الاتصال بين إكزونين أو ثلاث على مستوى الجين أما الآلآت التي تشفر سلاسل طويلة تسمح لنا بمعرفة الطرق المختلفة لترابط الإكزونات (Splicing Junctions) حيث أن أجزاءا من السلسلة تتطابق مثلا مع الأكزون الأول وجزئ آخر مع الاكسون الخامس والسابع وهكذا دواليك وبالتالي يمكننا معرفة أنواع البروتينات المنتجة.
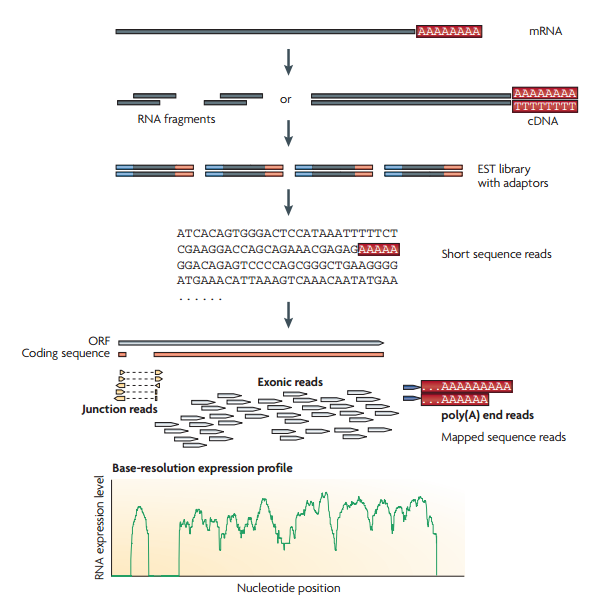
مخطط يبين مبدا تحضير ومطابقة السلاسل للجينوم (المصدر)
في بعض الآحيان يتم سَلْسَلة كل قطعة مرتين (من اليمين إلى اليسار و من اليسار إلى اليمين) للحصول على دقة أكثر بحيث أنه إذا كانت فيه بعض الأخطاء في القراءة الأولى (يمين-يسار) ربما لن تكون في القراءة الثانية(يسار-يمين), تسمى هذه الطريقة بالسلسلة ثنائية النهاية (Pair-end Sequencing) أما الطريقة العادية تسمى بالسلسلة أحادية النهاية (Single-end Sequencing)
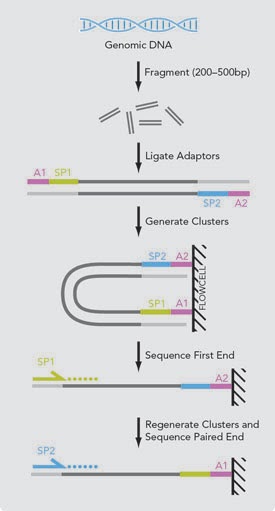
رسم توضيحي لمبدا السلسلة ثنائية النهاية (المصدر)
على الرغم من الشبه بين هذه التقنية و تقنية الـ Microarray من حيث الأهداف إلا أن هناك اختلاف كبيرا في الطرق الحسابية والاحصائية المستعملة في معالجة البيانات وتحديد الجينات التي تظهر تغيرا نسبيا في نسبة نشاطها. الجدول التالى يلخص بعض الاختلافات.
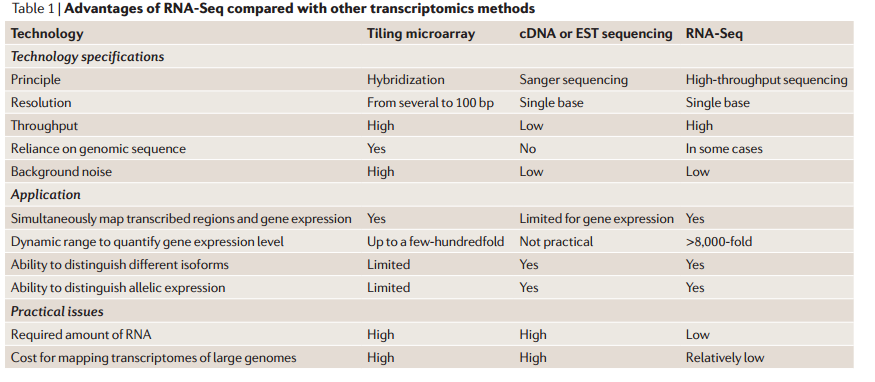
مقارنة بين تقنية الـ RNA-Seq والتقيات التي قبلها (المصدر)
بيانات السلسلة الخام (قوالب FASTQ )
بعد الإنتهاء من عملية السلسلة نتحصل على ملف من نوع FASTQ يحتوي على معلومات حول كل قطعة تمت سلسلتها (ملايين من السلاسل). تعتبر صيغة الـ FASTQ , الصيغة المتعارف عليها من أجل حفظ نتائج السَلْسلة مع إختلاف طفيف بين الآلات في طريقة حساب جودة التشفير. يتم كتابة معلومات عن كل قطعة cDNA في أربعة أسطر كالتالي:

1. السطر الأول (الرأس): يحدد بداية معلومات القطعة المشفرة ويبدأ دائما بالرمز "@" بعدها يحتوي على اسم القطعة (أو رقمها) لاتوجد صغية محددة لكن من الاحسن أن تحتوي على معلومات أكثر فمثلا في المثال الموضح في الصورة FC61KBDAAXX تمثل إسم الآلة. 7 يمثل رقم الممر (الصحيفة تكون مقسمة إلى ممرات), 2 يمثل رقم الرقاقة و 3478 و 5192 يمثلان إحداثيات قطعة الـ cDNA في الرقاقة. في حالة السلسلة ثنائية النهاية (Pair-end Sequencing) يمثل هنا الرقم 1 رقم السلسلة الأولى وفي الملف الذي يحتوي على معلومات السلسلة الثانية يكون لديها نفس السطر الاول لكن يكون رقم 2 في الأخير.
2. السطر الثاني: يحتوى على سِلسلة القطعة التي تم قراءتها
3. السطر الثالث: فاصل في العادة يكون الرمز +
4. السطر الرابع: يكون بنفس طول السلسلة في السطر الثاني. يمثل كل حرف منه جودة تشفير الجزيء القاعدي المقابل في السلسلة الموجودة في السطر الثاني مرمزة باستعمال شيفرة ASCII (حيث كما بينا في المقال السابق تقوم الآلة كل مرة بقراءة جزي قاعدي واحد ويقوم برنامج بتحليل الصورة لمعرفة نوع الجزيء القاعدي) .إذا كان 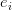 هو إحتمال الخطاء اثناء قرائة الجزي القاعدي
هو إحتمال الخطاء اثناء قرائة الجزي القاعدي 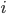 يمكننا حساب جودة التشفير بإستعمال إحدى الطريقتين:
يمكننا حساب جودة التشفير بإستعمال إحدى الطريقتين:
- قيمة الجودة Phred: أو Phred Quality Score ويطلق عليها أيضا اسم صيغة سانغر (Sanger Format) استعملت هذه الطريقة اثناء مشروع الجينوم البشري حيث يتم حساب الجودة Q كالتالي:
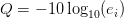
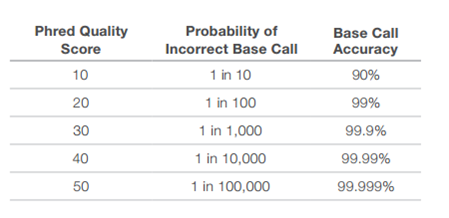
جدول يوضح قيم الجودة Phred
- قيمة الجودة Solexa/Illumina : تقوم آلات شركة Solexa/Illumia بحساب قيمة الجودة كالتالي:
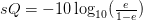
لهذا قبل الشروع في تحليل البيانات و تقصي جودة عملية السلسلة يجب أولا معرفة الصيغة المستعملة في حساب الجودة.
معالجة نتائج السَلْسلة:
تمثل نهاية المرحلة الأولى (السَلْسَلة) والحصول على ملف FASTQ بداية لمشوار طويل من تحليل البيانات ومحاولة استخراج اية معلومة يمكن ان تساعدنا في فهم البيولوجيا وراء هذه المعلومات. نظرا للعدد الهائل من التحاليل المختلفة التي يمكننا القيام بها سوف اقدم لمحة عن الخطوات الأكثر استعمالا وهي تقصي جودة المعلومات في ملف FASTQ ومطابقة قطع الـ cDNA المشفرة للجينوم ومعرفة المناطق النشطة وتحديد نسبة النشاط. اما التحاليل الاخرى (تحديد الطفرات SNP calling, مقارنة البيانات, تحديد التفاعلات الأيضية النشطة Metabolic Pathway analysis,... إلخ) سنحوال التطرق اليها في مقالات اخرى ان شاء الله.
1- تقصي جودة السلسلة: تعتبر هذه المرحلة اهم مرحلة يجب القيام بها لمعرفة مدى نجاح عملية السلسلة وهل كمية الـ cDNA المتحصل عليها كافية ونوع الأخطاء ان وجدت.
أولا في العادة نريد أن نعرف كم من قطعة تمت سلسلتها. مثلا إذا كان لدينا ملف "Tumor.1.1.fastq" يمكننا أن نعدعدد الأسطر التي تبدأ بالرمز "@" ففي نظام لينوكس مثلا يمكننا كتابة الأمر التالي:
grep -c "^@" Tumor.1.1.fastq 3066237
مثلا هنا لدينا أكثر من ثلاثة ملايين قطعة.
لتقصي جودة السلسلة يمكننا استعمال بعض البرامج المعروفة مثل fastqc الذي يوفر للمستعمل امكانية استعمال الواجهة أو استعمال البرنامج عن طريق اوامر الـ Shell. فمثلا اذا اردت تحليل ملف "Tumor.1.1fastq" يمكنك استعمال الأمر التالي اذا كان ملف
fastqc Tumor1.1.fastq
يقوم البرنامج بعدها بانتاج تقرير كامل على شكل HTML فمثلا في هذا المثال نلاحط متوسط الجودة بالنسبة لكل جزيء قاعدي, نلاحط أن طول كل قطعة 75 جزيء قاعدي وانه جودة السلسلة جيدة (  ) يعني نسبة خطا أقل من 0.001
) يعني نسبة خطا أقل من 0.001
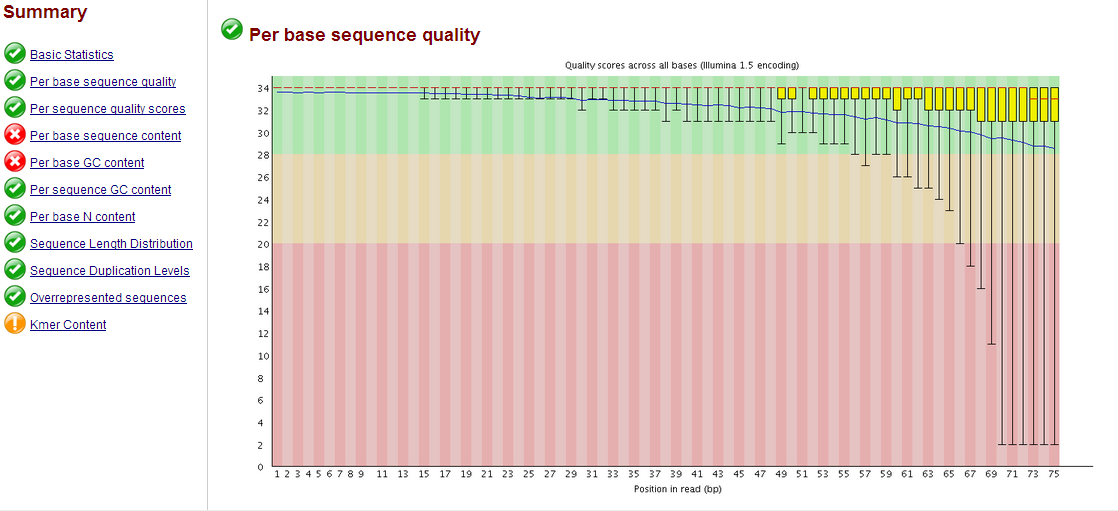
مثال عن مخطط نسبة جودة السلسلة
اذا كانت هناك بعض الاخطاء في عادة تكون في الأطراف (البداية أو النهاية) يمكن نزع الجزئ السيئ كما يمكن حذف القطع التي لا تتناسب مع الجودة المطلوبة.
2- تحديد موقع كل قطعة (Read Mapping) : هناك عدة برامج يمكن استعمالها لمطابقة قطع الـ cDNA مع الجينوم المرجعي (مثلا جينوم الانسان). من البرامج الأكثر استعمالا هناك BWA, Bowtie. لكن هذه الخوارزميات تعطيك معلومات أولية فقط عن عدد القطع التي تكن البرنامج من مطابتها و موقعها في الجينوم لكن لاتعطي تأخذ بعين الاعتبار الـ isoforms (او القطع التي لايمكن مطابقتها للسلسىة كاملة من الجينوم), برنامج Tophat يستغل نتائج Bowtie و يقوم بإيجاد هذه القطع.
لاستعمال برنامج tophat, يجب تثبيت برنامج Bowtie و Samtools. يجب أيضا أن تقوم بفهرسة الجينوم (Genome Indexing) الذي تريد اسعتماله باستعمال برنامج Bowtie أو تحميل جينوم مفهرس من موقع Bowtie. من أجل انشاء فهرس يجب أن يكون الجينوم على شكل ملف fasta ثم كتابة الأمر التالي:
bowtie2-build myGenome.fa myGenome_index
في حالتنا سمينا الفهرس hg19 (نسبة للنسخة الأخيرة لجينوم الانسان) وللحصول على دقة اكثر في المطابقة للجينات المعروفة يمكنك تحميل مواقع الجينات في الجينوم من موقع UCSC واختيار الحقول الموضحة في الصورة

توضيح كيفية تحميل معلومات حول موقع الجينات في الجينوم البشري من موقع UCSC
اذا فرضنا ان كل هذه الملفات في مجلد واحد (او في لينوكس يكنك صنع رابط افتراضي للملف لتفادي النسخ) يمكننا القيام بالمطابقة باستعمال Tophat كالتالي:
tophat2 –p 8 –o tumor1.1_thout --library-type fr-unstranded -G genesh19.gtf hg19 Tumor.1.1.fastq
بعد انتهاء العملية نتحصل على المفات التالية
| accepted_hits.bam |
يحتوي على السلاسل التي أمكن مطابقتها محفوظة بصيغة BAM |
| unmapped.bam |
يحتوي على السلاسل التي لم يمكن مطابقها محفوظة بصيغة BAM |
| deletions.bed |
يحتوي على قائمة السلاسل التي يحتمل ان تكون isoform |
| insertions.bed |
يحتوي على المواقع التي يحتمل ان تكون فيها طفرة اظافة جزيء قاعدي |
| junctions.bed |
يحتوي على المواقع التي يحتمل ان يكون فيها طفرة نزع جزيء قاعدي |
لحصول على بعض الاحصائيات على عدد السلاسل التي أمكن مطابقتها يمكنك استعمال Samtools
samtools flagstat accepted_hits.bam
لأن هناك احتمال أن يكون هناك تشابه كبير بين بعض السلاسل ومناطق عديدة من الجينوم, نريد أن يكون عدد السلاسل التي طوبقت للمكان واحد (Single Hit reads) من الجينوم كبيرا, لان مطابقة سلسلة لعدة أماكن يمثل غموض ولا يمكننا من معرفة المكان الحقيقي, لهذا من الأحسن تفقد نسبة السلاسل أحادية المطابقة يمكن حساب عدد السلاسل احادية المطابقة بالطريقة التالية:
samtools view -F 0x40 accepted_hits.bam | perl -F'\t' -ane 'print if(/NH\:i\:1[\t\n]/)' |wc -l
هذه فقط نظرة عابرة على بعض المعلومات التي يمكن معرفتها من أراد معلومات أكثر يمكن أن يراجع يطلع على صيغة ملفات SAM و BAM.
3- تقدير نسبة نشاط الجينات و الأيزوفورمات : تقدر نسبة نشاط موقع من الجينوم بكمية السلاسل التي تم مطابقتها للموقع. هناك طرق عديدة لحساب نسبة النشاط لكن من أكثرها استعمالا هي الـ RPKM اختصارا لـ Reads Per Kilobase of transcript per Million mapped reads اختارا هذه العبارة تعني, اذا استعملنا مليون كوحدة حساب العدد الاجمالي للسلاسل التي تم مطابقتها و اعتيرنا الكيلوبيز (1000 جزيء قاعدي) كوحدة لحساب طول الجين ماهي نسبة السلاسل التي تم مطابقتها لهذا الجين للعدد الاجمالي. عند تبسيط المعادلة يمكننا كتابتها كالتالي

بحيث يمثل :
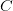 : عدد السلاسل التي تم مطابقتها لاكزونات الجين
: عدد السلاسل التي تم مطابقتها لاكزونات الجين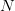 : العدد الاجمالي للسلاسل التي تم مطابقتها لكامل الجينوم
: العدد الاجمالي للسلاسل التي تم مطابقتها لكامل الجينوم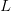 : طول الجين (عدد الجزيئات القاعدية)
: طول الجين (عدد الجزيئات القاعدية)
مثلا لو كان لدينا جين طوله L=1000 جزيء قاعدي وتم مطابقة C=2000 سلسلة له وكان العدد الاجمالي للسلاسل التي تمكننا من مطابقتها لكن انحاء الجينوم 8 ملايين يكون الـ 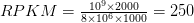
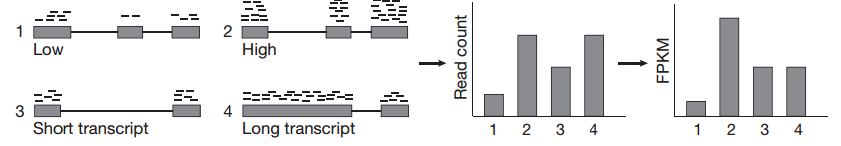
توضيح مبدا نسبة نشاط الجين (المصدر) (في الصورة لدينا FPKM لكن تقريبا نفس المبدا مع RPKM)
لحسن الحظ يمكن حساب هذه القيم باستعمال برامج كثيرة مثل Cufflinks
cufflinks -p 8 -o tumor1.1_clout tumor1.1_thout/accepted_hits.bam
بعدها نتحصل على الملفات التالية
| genes.fpkm_tracking |
نسبة نشاط الجينات |
| isoforms.fpkm_tracking |
نسبة نشاط الأيزوفرمات |
| skipped.gtf |
المناطق التي لم حسابها |
| transcripts.gtf |
نسبة نشاط كل النواسخ |
وفي الاخير أتمنى أن يكون هناك من استفاد من هذا المقال الذي هو موجه للمبتديين في المجال خاصة. يمكن بالطبع استعمال لغات برمجة كالـ R مثلا للقراءة الملفات والحصول على معلومات اكثر. يجب التنويه أنني لم اتطرق إلى كيفية مقارنة نشاط الجينات بين عينتين مختلفتين. يمكن القيام بهذا بمطابقة كل عينة على حدى ثم استعمال برنامج أمر Cuffdiff الذي هو جزء من Cufflinks. يمكن للقارئ المهتم أن يطلع على صفحة البرنامج للمزيد من التفاصيل.
وأنوه أيضا أنني لست متأكدا من كيفية ترجمة بعض المصطلحات مثل Pair-end Sequencing, Single-end Sequencing, Reads
رابط المقالة : المعلوماتية الحيوية بالعربية » استعمال طريقة RNA-Seq لدراسة نسبة نشاط الجينوم