السلام عليكم,
في الجزئين الاولين من هذه السلسلة (هنا وهنا) قمنا بشرح بعض المفاهيم الأساسية لمجال الـ Single-cell Sequencing وبينا كيفية الحصول على البيانات الاولية المتمثلة في مصفوفة التعبير الجيني. على الرغم من اننا استعملنا آلة Chromium 10x كمثال الا ان المبدا يبقى نفسه مع التقنيات الاخرى مع اختلاف في التفاصيل. في هذه المقالة سوف نحاول شرح اهم مراحل تحليل هذه البيانات ونقدم امثلة بلغة الآر على ذلك. في اسفل المقال وضعنا مجموعة من المصادر التي يمكن للقارىء المهتم الاطلاع عليها.
مقدمة
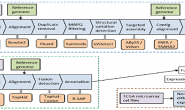
على الرغم من حداثة تقنيات الـ Single-cell sequencing الا انه تم تطوير العديد من الحُزم للتعامل مع هذه البيانات. مثلا في المخطط اسفله نلاحظ ان عدد الحُزم الخاصة بالـ Single-cell sequencing في مخزن Bioconductor فقط نما بسرعة مقارنة بعدد حُزم مجالات اقدم, كمجال الـ Meta-genomics. زد الى ذلك العديد من الحُزم الاخرى في مخزن CRAN و Github وحُزم بلغات اخرى كالبايثون وغيرها.
لهذ في بعض الاحيان يصعب على المبتدئين في المجال اختيار الحُزم التي يريدون استعمالها. في رايي, اذا كنت جديدا في المجال, من الاحسن البدأ باستعمال الحُزم الشائعة (يمكن الاطلاع على عدد الـ citation للورقة البحثية المرافقة للحزمة) ومن بعدها اذا صادفتك مشكلة لا تستطيع هذه الحُزم المشهورة حلها, يمكنك محاولة استعمال الحُزم الاقل شيوعا و التي ربما لن تجد العديد من الامثلة عن كيفية استعمالها. لحسن الحظ, قام الباحثون بوضع صفحة في Github تحتوي على قائمة بكل الحُزم المتوفرة يمكن الاطلاع عليها هنا.
في هذا الجدول, لخصنا قائمة ببعض الحزم الأكثر استعمالا والتي سوف نستعمل بعضها في هذا المقال.
| Seurat | من اكثر الحزم استعمالا, يمكن تحميلها من مخزن CRAN او من Github. يحتوي على مجموعة متكاملة من الدوال. |
| scater | يعتبر من اكثر حزم Bioconductor الاكثر استعمالا. يحتوي على العديد من الدوال من اجل التاكد من نوعية البيانات و انشاء الرسوم البيانية. |
| scran | يحتوي على مجموعة من الدوال تسمح لك بالتعامل بتحكم اكبر في طريقى تحليل بيانات الخلايا المفردة. يمكن تحميله من مخزن Bioconductor ايضا. |
| DropletUtils | تحتوي على مجموعة من الدوال للتعامل من البيانات الناتجة على عن الآلات التي تستعمل القطيرات (Droplets) كآلة 10x وغيرها. يمكن تحميل هذه الحزمة من مخرن Bioconductor . |
| SingleCellExperiment | تعرف فئة (Class) من نوع S4 لتخرين بيانات الخلايا المفردة. ترث من هذه الفئة كل حزم Biocondctor التي تتعامل مع بيانات الخلايا المفردة كـ scater و scran وغيرها. يمكن تحميلها من Bioconductor. |
مراحل تحليل البيانات
تمثل مصفوفة التعبير الجيني (count matrix) التي نتحصل عليها البيانات في شكلها الخام فقط ولا يمكن استعمالها مباشرة. يجب اولا التأكد من جودتها ونزع بيانات الخلايا والجينات رديئة النوعية وبعدها القيام بمجموعة من التحويلات و دمج لبيانات التجارب المختلفة و من ثم تحديد انواع الخلايا الموجودة في العينة وتحديد الجينات الواسمة (marker genes) لكل نوع من الخلايا. بعد تحديد انواع الخلايا, يمكننا القيام بدراسات اخرى كايجاد العلاقة النمائية (Developmental relationship) بين انواع الخلايا (اذا كنا مثلا ندرس ظاهرة نمائية) او تحديد انوع الخلايا الاكثر تأثرا بمرض من الامراض ... الخ.
في المخطط اسفله لخصنا اهم مراحل تحليل بيانات التعبير الجيني للخلايا المفردة (Single-cell RNA-Seq او اختصارا scRNA-seq).
من اجل الخروج من دائرة الشرح النظري للامور, سوف نقوم بتحميل بعض البيانات الحقيقية ونقوم بتحليلها وشرح الخطوات المبينة في المخطط شرحا نظريا وعملي لكي تعم الفائدة.
التعريف بالبيانات المستعملة
سوف نستعمل بيانات من ورقة بحثية نُشرت سنة 2018 في مجلة Nature communications والتي يحاول خلالها الباحثون استعمال تقنية الخلايا المفردة Chromium 10X لدراسة التنوع الخلوي للخلايا الطلائية (epithelial) و الخلايا الحشوية المتوسطة (mesenchymal) وتطور تعبيرهما الجيني اثناء مراحل التَخَلُق العضوي (organogenesis) لبنكرياس الفار خلال الايام الجنينية 14.5 , 12.5 و 17.5 (E14.5, E12.5 and E17.5). يأمل الباحثون من خلال هذه الدراسة ان يكتشفوا انواع خلايا جديدة لم يكن ممكن اكتشافها باستعمال الطرق الكلاسيكية وان يدرسوا التغيرات الجينية التي تحدث لكل نوع من الخلايا خلال هذه الفترة واكتشاف عوامل النسخ (Transcription factors) التي تساهم في هذه العملية, وهو شي صعب القيام به باستعمال RNA-Seq التقليدية.
تحميل البيانات
اذن لكي نحمل بيانات هذه الورقة البحثية نذهب الى نهاية الصفحة تقريبا وتحت عنوان Data availability سوف نجد رابط البيانات في مخزن GEO (Gene expression Omnibus) وهو الموقع المعتاد لوضع بيانات الـ NGS. في هذا المقال سوف نحمل جزءا فقط من البيانات الذي استعمل في الجزء الاول من الورقة البحثية.
تحضير البيانات و مساحة العمل
لكي يكون عملنا منظما من الاحسن ان تكون لدينا مساحة عمل منظمة. في العادة افضل استعمال حزمة template لقد قمت ببعض التعديلات للحزمة الاصلية لانها تحتوي على بعض المشاكل مؤخرا. في RStudio يمكنك تنصيب الحزمة كالتالي:
require("devtools") install_github("sirusb/template")
بعد التنصيب, نستعمل حزمة template لإنشاء مساحة عمل كالتالي:
require(devtools) require(template) dir.create("scRNASeq_example") setwd("scRNASeq_example/") new_project("ByrnesscRNASeq")
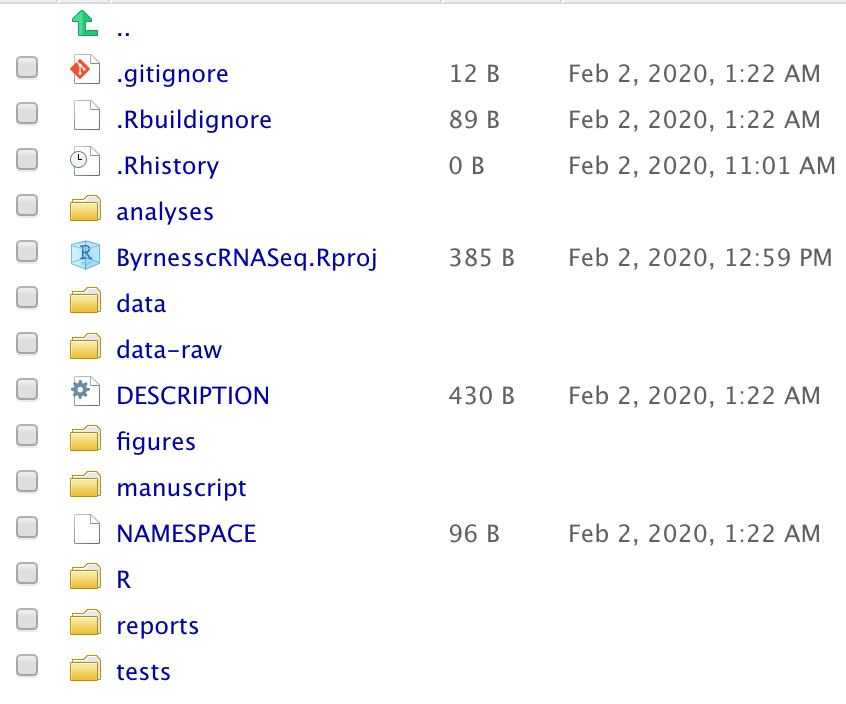
سوف تقوم الحزمة بانشاء مشروع جديد بإسم ByrnesscRNASeq يحتوي على الملفات المُبينة في الصورة اسفله. في هذا المقال سوف نقوم بكتابة الكود وحفظه في مجلد analyses و نضع البيانات التي حملناها من موقع GEO في مجلد data-raw و نحفظ البيانات التي ننشئها خلال تحليل البيانات في مجلد data و الصور في مجلد figures.
اذن نقوم بنسخ البيانات التي حملناها الى مجلد data-raw ونقوم بفك ضعطها.
mv ~/Downloads/GSE101099_RAW.tar ByrnesscRNASeq/data-raw tar -zxvf GSE101099_RAW.tar rm GSE101099_RAW.tar
>ls -1 GSE101099_RAW.tar GSM2699154_E14_B1.mtx.gz GSM2699154_E14_B1_barcodes.tsv.gz GSM2699154_E14_B1_genes.tsv.gz GSM2699155_E14_B2.mtx.gz GSM2699155_E14_B2_barcodes.tsv.gz GSM2699155_E14_B2_genes.tsv.gz GSM2699156_E12_B2.mtx.gz GSM2699156_E12_B2_barcodes.tsv.gz GSM2699156_E12_B2_genes.tsv.gz GSM2699157_E17_B2.mtx.gz GSM2699157_E17_B2_barcodes.tsv.gz GSM2699157_E17_B2_genes.tsv.gz
كل عينة تحتوي على ثلاث ملفات: ملف mtx.gz الذي يحتوي مصفوفة التعبير الجيني و ملف barcodes.tsv.gz الذي يحتوي على باركودات الخلايا وملف genes.tsv.gz الذي يحتوي على اسماء الجينات.
بما اننا سوف نستعمل حزمة Seurat لقراءة هذه البيانات, يجب ان نقوم ببعض التعديلات بجيث نصنع ملف لكل عينة و نغير اسماء الملفات في كل عينة الى : matrix.mtx, genes.tsv و barcodes.tsv. كما هو مبين في الكود اسفله
for smp in E14_B1 E14_B2 E12_B2 E17_B2 do mkdir -p $smp mv *${smp}*mtx.gz $smp/matrix.mtx mv *${smp}*genes.tsv.gz $smp/genes.tsv mv *${smp}*barcodes.tsv.gz $smp/barcodes.tsv done
الان ولقد جهزنا كل شيئ, فلنبدأ في تحليل هذه البيانات.
ضبط جودة البيانات (Quality control)
لنقم اذا بانشاء ملف ذو امتداد .rmd في مجلدanalyses نسميه مثلا analysis_workflow.Rmd. لكي نتبع نفس خطوات الورقة البحثية, نركز اولا على المرحلة الجنينية E14 لانها تعتبر الفترة الرئيسية التي يبدا فيها التنوع الخلوي خلال نمو البنكرياس. سوف نستعمل بصفة رئيسية حزمة Seurat و دالة Read10X لقرائة مصفوفة التعبير الجيني كما يلي:
E14_exprs <- list() E14_exprs[['batch1']] = Read10X("../data-raw/E14_B1/") E14_exprs[['batch2']] = Read10X("../data-raw/E14_B2/") lapply(E14_exprs,dim)
#$batch1 #[1] 27775 3849 #$batch2 #[1] 27775 5179
من الارجح ان الباحثين قاموا بسلسلة خلايا الـ E14 في دفعتين (batches) من اجل الحصول على عدد كبير من الخلايا. في الدفعة الأولى (Batch1) تم الحصول مبدايا على 3849 خلية و في الدفعة الثانية (Batch2) تم الحصول على 5179 خلية, اي مامجموعه 9028 خلية و تم حساب التعبير الجيني لـ 27775 جين في كلتا الدفعتين (بعض الجينات ربما ليس لها اي تعبير).
لكي نتعامل مع هذه المصفوفات بشكل اسهل, نحولها الى Seurat objects كالتالي:
E14_sobjs = list() for(batch in names(E14_exprs)){ E14_sobjs[[batch]] = CreateSeuratObject(counts = E14_exprs[[batch]],project = batch) } E14_sobjs
# $batch1 # An object of class Seurat # 27775 features across 3849 samples within 1 assay # Active assay: RNA (27775 features) # $batch2 # An object of class Seurat # 27775 features across 5179 samples within 1 assay # Active assay: RNA (27775 features)
قبل استعمال هذه البيانات يجب علينا اولا ان ننزع الخلايا التي لم نتمكن من سلسلة عدد كافي من جيناتها وان ننزع ايضا الجينات التي لم يتم رصدها في عدد كافي من الخلايا لانها لا تعطينا معلومات مفيدة و ممكن ان تعطينا نتائج خاطئة.
تتميز الخلايا ذات النوعية الرديئة بعدة خصائص من بينها:
- وجود عدد كبير من السلاسل القادمة من الميتوكوندريا وذلك لتمزق الخلية.
- تحتوي على عدد قليل من الجينات المعبر عنها بسبب عدم فاعلية عملية النسخ العكسي او الـ PCR.
- ليست خلايا حقيقية بل قطيرات تحتوي على بعض الـ RNA الطائفة في السائل.
في هذه البيانات, قام الباحثون بنزع معلومات جينات المايتوكوندريا (كما نرى في الكود اسفله), لهذا من الصعب معرفة ذلك (اذا اردنا ان نعرف ذلك يجب ان نحمل ملفات fastq الاصلية ونعيد مطابقتها).
for(batch in names(E14_sobjs)){ E14_sobjs[[batch]][["percent.mt"]] <- PercentageFeatureSet(E14_sobjs[[batch]], pattern = "^mt-") } quantile(E14_sobjs$batch1$percent.mt)
# 0% 25% 50% 75% 100% # 0 0 0 0 0
بما ان هذه البيانات لاتعطينا معلومات حول الميتوكوندريا سوف نتاكد من عدم وجود خلايا ذات عدد قليل من الجينات المعبر عنها. عند استعمال حزمة Seurat تقوم اوتوماتيكيا بحساب عدد الجينات المعبر عنها في كل خلية وتضع ذلك في جدول meta.data في كل كائن كما هو مبين هنا في بيانات batch1. كل سطر يعطينا معلومات عن خلية واحدة وتظاف حقول جديدة لهذا الجدول كلما قمنا بتحليلات ايضافية, حيث يمنكننا ان نضيف حقل جديد, كما فعلنا مع المايتوكوندريا باضافتنا حقل percent.mt.
head(E14_sobjs$batch1@meta.data)
# orig.ident nCount_RNA nFeature_RNA percent.mt # AAACATACATACCG batch1 8584 2833 0 # AAACATACCAAGCT batch1 3302 1741 0 # AAACATACCGCTAA batch1 5301 2374 0 # AAACATTGAAACAG batch1 17067 4280 0 # AAACCGTGGCAGTT batch1 4842 2199 0 # AAACCGTGTAGACC batch1 2318 1133 0
في هذا الجدول لدينا مبدئيا الحقول التالية:
orig.ident: يحتوى على اسم العينة (batch1 او batch2).nCount_RNA: يحسب عدد سلاسل الـ RNA القادمة من هذه الخلية. في حالة Chromium 10x يمثل عدد الـ UMI.nFeature_RNA: يمثل عدد الجينات المعبر عنها. يعتبر الجين معبر عنه اذا تحصلنا على الاقل على UMI واحد منه.percent.mt: ويمثل نسبة الـ UMI الناتجة من جينات المايتوكوندريا. هذا الحقل اضفناه يدويا في الكود فوق.
اذن سوف نستعمل بيانات الحقل nFeature_RNA لمعرفة توزيعها الاحصائي ومعرفة الى الحد (Threshold) الذي نستعمله لتقرير ما اذا كانت الخلية تعبر عدد قليل من الجينات ام لا.
p1 = VlnPlot(E14_sobjs$batch1, features = c("nFeature_RNA"), ncol = 1,pt.size = 0.2,cols = "#0073C2FF") + geom_hline(yintercept = 500,color="red") p2 = VlnPlot(E14_sobjs$batch2, features = c("nFeature_RNA"), ncol = 1,pt.size = 0.2,cols = "#868686FF") + geom_hline(yintercept = 500,color="red") CombinePlots(list(p1,p2),ncol = 2)
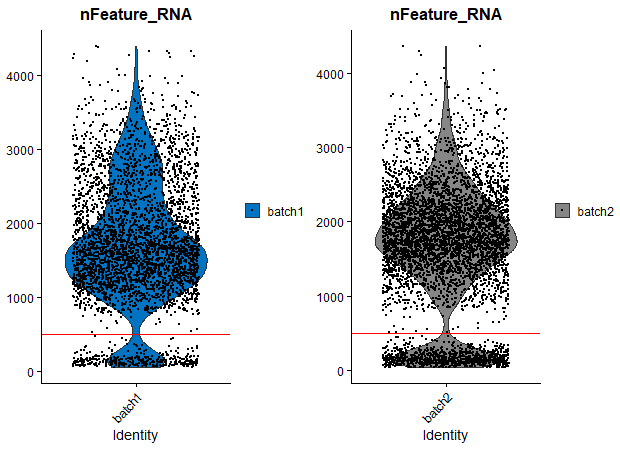
نلاحظ اننا تمكنا من قياس التعبير الجيني لاكثر من 1000 جين في اغلبية الخلايا, الا ان هناك عدد من الخلايا (تحت الخط الاحمر) لديها عدد قليل من الجينات المعبر عنها (اقل من 500 جين).
نقوم اذا بحذف هذه الخلايا كالتالي:
for(batch in names(E14_sobjs)){ E14_sobjs[[batch]] <- subset(E14_sobjs[[batch]], nFeature_RNA >500) } E14_sobjs
$batch1 An object of class Seurat 27775 features across 3495 samples within 1 assay Active assay: RNA (27775 features) $batch2 An object of class Seurat 27775 features across 4309 samples within 1 assay Active assay: RNA (27775 features) E14_sobjs
نلاحظ اننا قمنا بنزع 354 خلية من batch1 و 870 خلية من batch2 وتبقى لنا 3495 و 4309 خلية.
تسوية البيانات (Normalization)
الاصل ان تكون قيم التعبير الجيني المتحصل عليها في مصفوفة التعبير الجيني (count matrix) متناسبة طرديا مع عدد نسخ الـ mRNA لكن جين في كل خلية. مثلا, اذا كان لدى الخلية c1 ضعف عدد نسخ الـ mRNA للجين g1 مقارنة بالخلية c2, الاصل نرى ذلك في مصفوفة التعبير الجيني. لكن كما هو الحال عند التعامل مع البيانات التجريبية بصفة عامة, لا يمكننا استعمال البيانات الخام مباشرة لانها لا تخلوا من التشويشات التقنية (Technical variation) الناتجة عن التباين في فاعلية كل مرحلة تجريبية في كل قطيرة (Droplet) كالـ Reverse transcription, PCR وغيرها التي تؤدي الى انحياز في قيمة التعبير الجيني (كما هو مبين في الصورة اسفله). لهذا يجب علينا استعمال بعض الطرق الاحصائية لتسوية البيانات.
خلال استعمال تقنية الـ RNA-Seq التقليدية اذا كان لدينا عمق سَلسَلة كافي ( 30 الى 40 مليون سِلسِلة) وكان التعبير الجيني لجين من الجينات معدوم فيمكننا ان نجزم بنسبة عالية من الثقة ان كمية الـ mRNA لهذا الجين قليلة جدا او معدومة في هذه العينة. خلافا للـ RNA-Seq التقليدية تتميز تقنية الـ scRNA-Seq بفاعلية التقاط ونسخ عكسي للـ mRNA (ممثلة بالمتغير Fj في الصورة ) بنسبة تتراوح مابين 10% الى 20% لهذا فوجود تعبير جيني معدوم في مصفوفة التعبير الجيني يحتمل تفسيران: إما ان الخلية لا تعبر عن هذا الجين او اننا لم نستطع التقاطه, تسمى هذه الظاهرة بالـ drop-out event. لهذا تتميز مصفوفة التعبير الجيني للـ scRNA-Seq بكونها مليئة بالاصفار ( Sparse matrix).

مثلا في الصورة أعلاه قام الباحثون بتحليل التعبير الجيني لخليتين متطابقتين لكن نلاحظ ان هناك بعض الجينات يتم التقاطها في خلية وليس في الاخرى ذلك لاسباب تقنية وبيولوجية كالانفجار التعبيري (Transcriptional burst).
في تسوية البيانات أيضا نريد ان لا تكون كمية التعبير الجيني متناسبة طرديا مع عمق السَلسَلة (Sequencing depth) للخلية لانه عند المقارنة بين الخلايا لايمكننا التقرير هل هذا اختلاف بيولوجي ام فقط تقني. فمثلا في البيانات نلاحظ هذا المشكل, كما هو مبين هنا:
smoothScatter(E14_sobjs$batch1@meta.data$nCount_RNA, E14_sobjs$batch1@meta.data$nFeature_RNA, xlab = "Sequencing depth", ylab = "number of Detected genes")
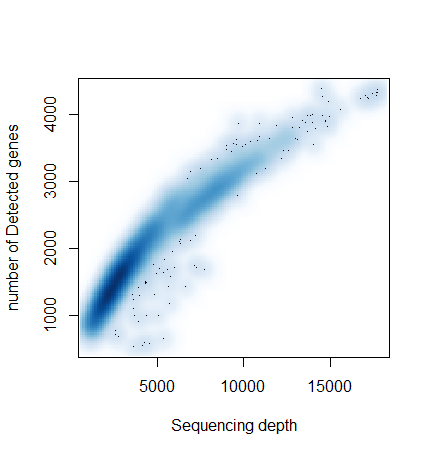
الطريقة الكلاسيكية للقضاء على العلاقة بين قيمة التعبير الجيني وعمق السَلسَة (Sequencing depth) هي قسمة التعبير الجيني للجينات في كل خلية على عدد العدد الكلي للسلاسل الُمتحصل عليها في الخلية (library size). نقوم في العادة بظرب هذا العدد بمليون او عشرة الاف لكي لا نتحصل على ارقام صغيرة جدا (كما هو موضح في المعادلة اسفله). تسمى هذه الطريقة في التسوية بالـ CPM (counts per million) Normalization. في العادة نقوم بتحويلها الى  لتتقليص من تاثير القيم الشاذة على الحسابات.
لتتقليص من تاثير القيم الشاذة على الحسابات.
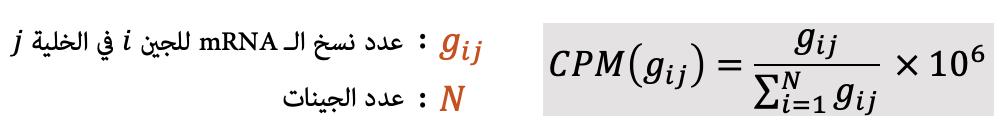
في Seurat يمكننا حساب ذلك باستعمال دالة NormalizeData و تحديد الطريقة normalization.method = "LogNormalize"
E14_sobjs = lapply(E14_sobjs, NormalizeData,normalization.method = "LogNormalize")
على الرغم من الاستعمال الواسع لطريقة الـ CPM الا انها مشكلتها تتمثل في انها تقوم باستعمال معامل مقياسي (scaling factor) واحد (  ) على كل جينات الخلية الواحدة. لاحظ الباحثون انه لو قسمنا الجينات على حسب تعبيرها الى مجمعوعات (هنا ستة, الصور A و B اسفله) سوف تتمكن طريقة الـ CPM من تسوية بيانات فقط الجينات العالية التعبير الجيني (ليس لها العديد من الـ droupouts) لكن تفشل في تسوية التعبير الجيني للخلايا ضعيفة التعبير (كما هو مبين اسفله في الصورة C).
) على كل جينات الخلية الواحدة. لاحظ الباحثون انه لو قسمنا الجينات على حسب تعبيرها الى مجمعوعات (هنا ستة, الصور A و B اسفله) سوف تتمكن طريقة الـ CPM من تسوية بيانات فقط الجينات العالية التعبير الجيني (ليس لها العديد من الـ droupouts) لكن تفشل في تسوية التعبير الجيني للخلايا ضعيفة التعبير (كما هو مبين اسفله في الصورة C).

لهذا اقترح بعض الباحثون ان تتم تسوية التعبير الجيني على مستوى مجموعات الجينات عوض الخلايا كما هو في الـ CPM. من بين الطرق المقترحة حديثا هي طريقة SCTransform التي تمثل العلاقة بين متوسط عدد الـ UMI المتحصل عليه لكل جين و مدى عمق سلسلته في مجموع الخلايا على شكل نموذج الخطي العالم (Generalized Linear Model) للتوزيع الـ Negative Binomial كالتالي:
 .
.
بحيث يمثل  عدد UMI الجين
عدد UMI الجين 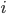 في الخلية
في الخلية  و
و  يمثل القيمة الابتدائية و
يمثل القيمة الابتدائية و  يمثل نسبة تاثير عمق السلسلة. تقوم SCTransform بحساب معيار التشتت و في الاخير تحسب Pearson residual كالتالي
يمثل نسبة تاثير عمق السلسلة. تقوم SCTransform بحساب معيار التشتت و في الاخير تحسب Pearson residual كالتالي  لتسعمله بعد ذلك لتسوية التعبير الجيني. في الصورة اسفله اقتباس للكود الذي يفعل ذلك في حزمة
لتسعمله بعد ذلك لتسوية التعبير الجيني. في الصورة اسفله اقتباس للكود الذي يفعل ذلك في حزمة sctransform.
في Seurat يمكننا استعمال طريقة SCTransform كالتالي:
E14_sobjs = lapply(E14_sobjs, SCTransform, verbose = TRUE)
اذا كنت مهتما اكثر بهذا النموذج يمكنك الاطلاع على نتائجه باستعمال الامر التالي:
sctransform::plot_model_pars(E14_sobjs$batch1@assays$SCT@misc$vst.out) + theme_classic()

يمكننا المقارنة بين فاعلية الـ CPM Normalization و SCT normalization باستعمال دالة plotRLE من حزمة التي تقوم بحساب الـ relative log expression (RLE) للجينات كل خلية. اذا كانت تسوية البيانات جيدة نتوقع ان يكون متوسط الـ RLE في كل خلية حوالي 0.
norm_plots <- list() i=1 for(batch in names(E14_sobjs)){ sce = Seurat::as.SingleCellExperiment(E14_sobjs[[batch]], assay="RNA") rs1= rowSums(GetAssayData(object = E14_sobjs[[batch]], slot = "counts",assay = "RNA") > 0) reliable = 0.1 * ncol(E14_sobjs[[batch]]) ttl = paste0("LogNormalizion (",batch,")") norm_plots[[i]] = plotRLE(sce[rs1>reliable,],exprs_values = "logcounts",colour_by = "orig.ident") + ggtitle(ttl) i=i+1 sce2 = Seurat::as.SingleCellExperiment(E14_sobjs[[batch]],assay="SCT") rs= rowSums(GetAssayData(object = E14_sobjs[[batch]], slot = "counts",assay = "SCT") > 0) ttl = paste0("SCT normalization (",batch,")") norm_plots[[i]] = plotRLE(sce2[rs>reliable,],exprs_values = "logcounts",colour_by = "orig.ident") + ggtitle(ttl) i=i+1 } CombinePlots(norm_plots,ncol = 2)
في حالة هذه البيانات نلاحظ ان طريقة الـ SCT تعطينا نتائج جيدة (الصورة اسفله).

دمج العينتين
كما اشرنا سابقا, قام الباحثون بالحصول على بيانات E14 على دفعتين, لكي نقوم بتحليل هذه البيانات يجب علينا ان ندمجها معا. المشكلة التي يمكن ان تواجهنا هي وجود تغيرات تقنية خاصة بظروف تحضير كل عينة او مايسمى يالـ Batch effect.
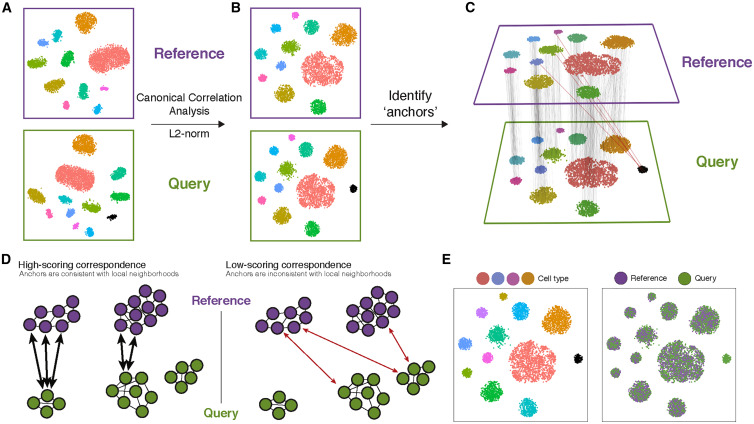
هناك العديد من الطرق التي تمكننا من تقليص الـ Batch effect في حالتنا هذه سوف نستعمل الطريقة المتوفرة في حزمة Seurat و التي تستعمل طريقة الـ Canonical Correlation Analysis. اولا يجب ان نختار الجينات التي لاتمثل Bach effect التي يمكن استعمالها للدمج العينتين. هنا نختار 3000 جين باستمعمال دالة SelectIntegrationFeatures بعدها نستعمل دالة PrepSCTIntegration للتحضير البيانات للدمج, في الاخير ننادي دالتي FindIntegrationAnchors و IntegrateData لاكمال عملية الدمج (كما هو مبين في الكود اسفله). لمزيد من المعلومات يمكن الاطلاع على تفاصيل هذه الدوال.
options(future.globals.maxSize=5000*1024^2) E14.features <- SelectIntegrationFeatures(object.list = E14_sobjs, nfeatures = 3000) E14_sobjs <- PrepSCTIntegration(object.list = E14_sobjs, anchor.features = E14.features, verbose = FALSE) E14.anchors <- FindIntegrationAnchors(object.list = E14_sobjs, normalization.method = "SCT", anchor.features = E14.features, verbose = FALSE) E14.integrated <- IntegrateData(anchorset = E14.anchors, normalization.method = "SCT", verbose = FALSE) E14.integrated
# An object of class Seurat # 46884 features across 7804 samples within 3 assays # Active assay: integrated (3000 features) # 2 other assays present: RNA, SCT
تقليص الابعاد (Dimension reduction)
بعدما دمجنا البيانات نريد ان نمثلها باستعمال طرق عرض البيانات المرئية لكي تُسهل علينا عملية تفسيرها و استنتاج بعض العلاقة بسهولة. مثلا في حالة البيانات التي لدينا تتكون مصفوفة التعبير الجيني من 46884 جين و 7804 خلية وهو حجم كبير ويصعب تمثيلها حتى باستعمال الـ Heatmap.
لكي نستطيع تمثيل البيانات اذن يجب علينا ان نقلل من حجم البيانات ونختار فقط البيانات المفيدة. نلاحظ اولا ان عدد 46884 جين عدد كبير جدا ومن المستحيل ان تكون كل هذه الجينات مهمة, فبعضها ذو تعبير ضعيف وبعضها لا تتغير قيمته بين الخلايا. لهذا فنحن نريد اختيار الجينات التي يكون معبر عنها فقط في مجموعة من الخلايا لانها تمكننا من اكتشاف المجموعات المختلفة من الخلايا. تسمى هذه الجينات الـ Highly Variable genes او HVG اختصارا. في Seurat يمكننا اكتشاف هذه الجينات باستعمال الدالة FindVariableFeatures. مثلا في الكود اسفله قمنا باختيار الـ 2000 جين الاكثر تغيرا في بياناتنا و كتبنا اسماء العشر جينات الاولى.
E14.integrated <- FindVariableFeatures(E14.integrated, selection.method = "vst", nfeatures = 2000) # Identify the 10 most highly variable genes top10 <- head(VariableFeatures(E14.integrated), 10) # plot variable features with and without labels plot1 <- VariableFeaturePlot(E14.integrated) plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE) plot2

طريقة تحليل المكونات الاساسية (PCA)
بعدما وجدنا الجينات المتغيرة يمكننا استعمالها في اكتشاف المجموعات, يمكننا استعمال طريقة الـ Heatmap لكن حجم هذه البيانات مازال كبيرا 2000 * 7804. اذن يجب علينا تخفيض حجم هذه البيانات لكي نستطيع تمثيلها في فضاء ذو ابعاد اقل (ثنائي او ثلاثي الابعاد), و من بين الطرق الشائعة في هذا المجال هي طريقة تحليل المكونات الاساسية (Principal Component Analysis) او PCA اختصارا. تسمح لنا PCA بعملية تحويل للمَعلم الفضائي بحيث تتمكن من التقاط اغلبية التبابين المتواجد في البيانات في عدد قليل من محاور المعلم الفضائي (في العادة المحاور الـ 2 او الـ 3 الاولى تلتقط اغلبية التباينات الموجودة).

في Seurat يمكننا القيام بعملية الـ PCA بكل بساطة باستعمال دالة RunPCA.
E14.integrated <- ScaleData(E14.integrated, verbose = FALSE) E14.integrated <- RunPCA(E14.integrated, verbose = FALSE)
يقوم Seurat بحساب الـ 30 عنصر اساسي الاولى من الـ PCA1 (يمكننا تغير ذلك). نريد اولا ان نعرف ماهي نسبة التباين (variability) التي تم تلخيصها في كل من هذه العناصر الاساسية. في العادة ناخد العناصر التي تمثل مامجموعه 80% من التباين. يمكننا معرفة ذلك باستعمال دلة ElbowPlot. بالاظافة الى ذلك يمكننا استعمال دلة DimPlot رسم العنصرين الاولين (او غيرهما) كالتالي:
p1 = ElbowPlot(E14.integrated) p2 = DimPlot(E14.integrated,reduction = "pca",group.by = "orig.ident") CombinePlots(list(p1,p2))
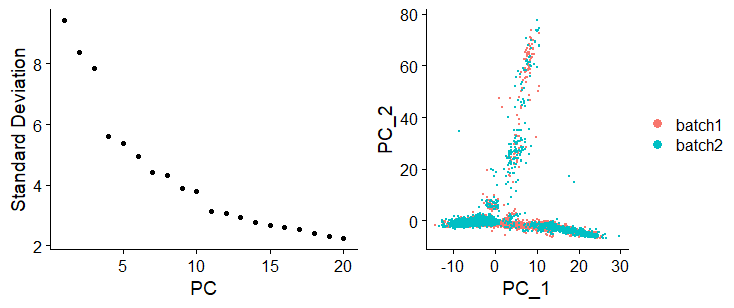
نلاحظ ان الثلاثة العناصر الأساسية الأولى مجتمعة لا تلخص الا حوالي 25% من التباين في البيانات مما يعني اننا بحاجة الى استعمال طرق اخرى كطريقة t-SNE التي تاخذ مجموعة من العناصر الاساسية وتلخصها في فضاء ثنائي الابعاد.
إختيار العناصر الأساسية
هناك طريقتين لاختيار العناصر الأساسية:
- الطريقة المرئية: هذه الطريقة تعتمد على الإختيار اليدوي, نقوم خلالها بعرض الجينات و الخلايا الأكثر تغيرا في كل عنصر أساسي ونختار العناصر الأساسية التي تمكننا من الحصول على مجموعتين (الكود اسفله):
DimHeatmap(E14.integrated, dims = 1:12, cells = 1000, balanced = TRUE)
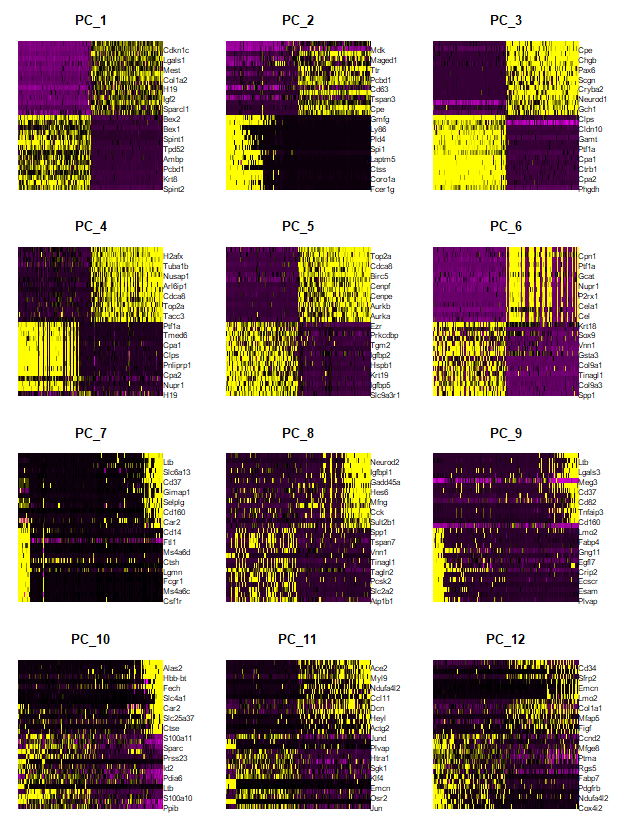
نلاحظ هنا ان بعض العناصر الاساسية (PC7 و PC9) لاتعطينا مجموعات واضحة, يمكننا اذن حذفهما.
- الطريقة الإحصائية: هذه الطريقة اكثر صرامة لانها تعطينا p-value يمكننا من خلالها تقرير ماهي العناصر الاساسية التي يمكننا استعمالها. تستعمل هذه الطريقة طريقة Jackstraw (لن اشرحها هنا). يمكننا استعمالها كالتالي :
E14.integrated <- JackStraw(E14.integrated, num.replicate = 100) E14.integrated <- ScoreJackStraw(E14.integrated, dims = 1:20) JackStrawPlot(E14.integrated, dims = 1:20)
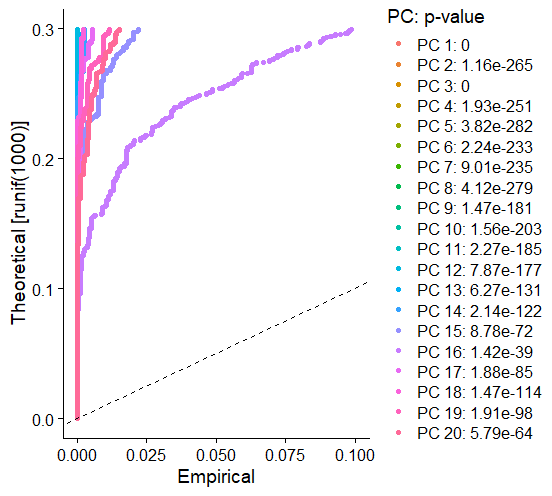
يمكن ان نلاحظ هنا ان العناصر الأساسية العشرين الأولى كلها مهمة ويمكننا استعمالها.
استعمال t-SNE او UMAP
هناك عدة طرق لتلخيص مجموعة من العناصر الاساسية في فضاء صغير, من اشهرها t-SNE و UMAP. يمكن استعمال دالة RunTSNE او RunUMAP للقيام بذلك. سوف نقوم اذن باستعمال t-SNE ونطلب منها ان تستعمل العشرين عنصر اساسي الأولى.
E14.integrated <- RunTSNE(E14.integrated, dims = 1:20) DimPlot(E14.integrated, group.by = "orig.ident",reduction = "tsne",cols=c("#0073C2FF", "#EFC000FF"))

نلاحظ انه تقريبا كل انواع الخلايا في العينة الثانية تم رصدها في العينة الاولى الا ان هناك مجموعة من الخلايا في العينة الثانية تم رصد عدد اكبر منها, هذا ربما ناتج عن الاختلاف قطع العينات. ربما في العينة قطعنا مساحة اكبر قليلا. يمكن ان يكون هذا الاختلاف ايضا ناتج عن عملية ضبط نوعية البيانات التي قمنا بها سابقا. لكن لن يؤثر هذا كثيرا على النتائج لاننا لسنا بصدد المقارنة بين العينتين.
تجميع الخلايا
بعد هذا كله يمكننا القيام بعملية التجميع (Clustering). هناك العديد من الطرق لن اتطرق لها هنا, لكن من اكثرها استخداما هي طريقة Louvain مع الـ K-nearest neighbors. يمكننا القيام بذلك كالتالي:
E14.integrated <- FindNeighbors(E14.integrated, reduction = "pca", dims = 1:20) E14.integrated <- FindClusters(E14.integrated, resolution = 0.5) DimPlot(E14.integrated, reduction = "tsne", label = TRUE,label.size = 5,pt.size = 0.7)

اكتشاف الـ markers
بعدما تحصلنا على القائمة الأولية لمجموعات الخلايا نريد ان نتعرف على هوية كل مجموعة من الخلايا. يكننا القيام بذلك عن طريق اكتشاف الجينات المعبر عنها حصريا في كل مجموعة. يمكننا القيام بذلك باستعمال دلة FindAllMarkers .
markers <- FindAllMarkers(E14.integrated,only.pos = TRUE)
Seurat يوفر العديد من الدوال من اجل عرض التعبير للجينات التي تهمنا او الجينات بعض الـ Markers كدالة VlnPlot و DotPlot وغيرها. مثلا هنا نعرض التعبير الجيني لبعض الجينات المبينة في الورقة البحثية باستعمال VlnPlot
DefaultAssay(E14.integrated) = "RNA" VlnPlot(E14.integrated, c("Cdh1","Chga","Rac2","Pecam1"),ncol = 2,pt.size = 0.1)
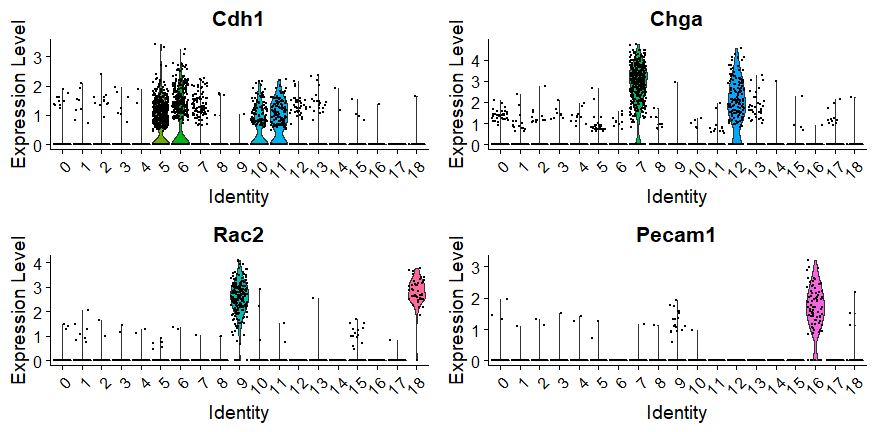
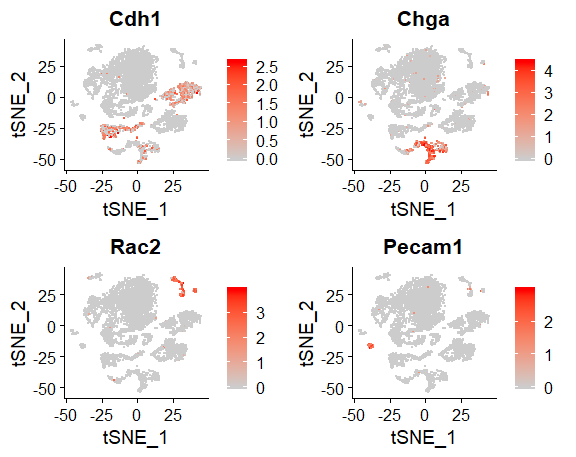
يمكننا ايضا عرض التعبير الجيني مباشرة في الـ t-SNE باستعمال دلة FeaturePlot كالتالي:
FeaturePlot(E14.integrated, features = c("Cdh1","Chga","Rac2","Pecam1"), ncol = 2, pt.size = 0.5, cols = c('grey80','red') ,max.cutoff = "q99")
الخلاصة
في هذا المقال الطويل سنتوقف هنا على الرغم من وجود العديد من التفاصيل والتقنيات التي يمكننا استعمالها, لكن الهدف من هذا المقال هو التعريف بالخطوط العريضة لتحليل بيانات الخلايا المفردة. يمكن للقارى المهتم ان يطلع على المصادر المرفقة بالمقال.
مصادر
- https://osca.bioconductor.org/
- https://scrnaseq-course.cog.sanger.ac.uk/website/index.html
- https://kkorthauer.org/fungeno2019/
- https://github.com/seandavi/awesome-single-cell
- L. Lun, A.T., Bach, K. & Marioni, J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol17, 75 (2016)
- Zhang, M.J., Ntranos, V. & Tse, D. Determining sequencing depth in a single-cell RNA-seq experiment. Nat Commun11, 774 (2020).
- Haghverdi, L., Lun, A., Morgan, M. et al. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol36, 421–427 (2018).
- Butler, A., Hoffman, P., Smibert, P. et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol36, 411–420 (2018).
- un, A., Riesenfeld, S., Andrews, T. et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol20, 63 (2019).
- Vallejos, C., Risso, D., Scialdone, A. et al. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat Methods14, 565–571 (2017).
- Tian, L., Dong, X., Freytag, S. et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat Methods16, 479–487 (2019).
- Lähnemann, D., Köster, J., Szczurek, E. et al. Eleven grand challenges in single-cell data science. Genome Biol21, 31 (2020).
رابط المقالة : المعلوماتية الحيوية بالعربية » مدخل الى مجال تحليل الخلايا المفردة ( الجزء الثالث)